Zespół XXY - Zespół Klinefeltera

Spis treści
Zespół Klinefeltera- informacje ogólne
Zespół Klinefeltera jest to zespół genetycznych wad wrodzonych, spowodowany obecnością u mężczyzn przynajmniej jednego, dodatkowego (nadliczbowego) chromosomu płci X w części lub we wszystkich komórkach organizmu. Standardowo człowiek posiada 22 pary chromosomów, które nie różnią się u osobników różnych płci (tzw. autosomy) oraz 2 chromosomy płci – determinujące płeć męską XY lub płeć żeńską XX. W Zespole Klinefeltera natomiast, poza standardową parą chromosomów płci mężczyzny XY, występuje dodatkowy, co najmniej jeden chromosom X, prowadząc do powstania tzw. trisomii chromosomów płci, np. XXY.
Częstość występowania Zespołu Klinefeltera jest zależna od rodzaju stwierdzonej ilości chromosomów płci. Postać wyrażana zapisem XXY jest zarazem najbardziej powszechną ze wszystkich aberracją chromosomów płci a jej częstotliwość określa się na poziomie 1:1500 – 1:5000 żywo urodzonych chłopców. Pojawienie się swoistych objawów jest zależne od wieku i charakteryzują się zróżnicowaniem, dlatego też rozpoznanie choroby następuje zazwyczaj w okresie dojrzewania chłopca.
Inne opisane postacie Zespołu Klinefeltera, występujące z mniejszą częstotliwością to:
- 48, XXYY: częstotliwość 1:17 000 – 1:50 000
- 48, XXXY: częstotliwość 1:17 000 – 1:50 000
- 49, XXXXY: częstotliwość 1:85 000 do 1:100 000
- 47, XXY/46, XY: mozaicyzm, bardzo rzadki obejmujący 10% wszystkich przypadków zespołu Klinefeltera
- 47 XXY/46 XX: mozaicyzm: bardzo rzadki, do 2016 roku opisano tylko 7 przypadków w piśmiennictwie medycznym

Diagnostyka zespołu Klinefeltera
W prenatalnej diagnostyce ilości chromosomów płci wykorzystuje się zarówno nieinwazyjne, jak i inwazyjne badania prenatalne. Z nieinwazyjnych badań prenatalnych najbardziej użyteczne są testy NIPT, które z bardzo dużym prawdopodobieństwem oceniają ryzyko wystąpienia zespołu Klinefeltera. Należy pamiętać, że badania NIPT to badania nieinwazyjne, i ich wynik nie zawiera ostatecznego rozpoznania. Prenatalnie potwierdzenie liczby chromosomów płci możemy uzyskać jedynie na drodze prenatalnych badań inwazyjnych (np. amniopunkcja).
Jeśli tego typu badania nie były wykonywane, to po urodzeniu dziecka, na podstawie zaobserwowanych objawów lekarz prowadzący może skierować pacjenta na badanie cytogenetyczne (kariotyp) w celu ustalenia liczby chromosomów. Jednakże, ze względu na opisane poniżej zróżnicowanie objawów, wykrycie zespołu Klinefeltera może nastąpić dopiero w okresie dojrzewania dziecka.
Podstawy genetyczne zespołu Klinefeltera
Zespół Klinefeltera oraz jego różne warianty nie jest dziedziczny. Do powstawania zespołu dochodzi przypadkowo w trakcie procesu dojrzewania komórek rozrodczych – komórek jajowych oraz plemników, a dokładniej w efekcie nie rozejścia się (nondysjunkcja) chromosomów homologicznych w trakcie ich dojrzewania (proces gametogenezy). Chromosomy homologiczne są to chromosomy o takiej samej budowie (wielkość i kształt), występujące w komórkach ciała w parach, po jednym chromosomie pochodzącym od matki i ojca. W wyniku nieprawidłowego rozejścia się chromosomów, do jednej z komórek rozrodczych trafia jedna lub więcej nadmiarowych kopii chromosomu X.
W większości przypadków do powstania zespołu dochodzi w wyniku nieprawidłowego dojrzewania plemnika (~50% przypadków) a badania populacyjne wykazały prawdopodobną zależność między wzrostem wieku przyszłego ojca a zwiększeniem ryzyka wystąpienia nieprawidłowości. Mozaicyzm 46,XY/47,XXY również nie jest dziedziczny, powstaje w wyniku nieprawidłowości podziałów komórkowych w trakcie wczesnego rozwoju zarodka. W wyniku czego część komórek ciała zawiera prawidłową ilość chromosomów X oraz Y (46, XY) a pozostałe komórki nadmiarowy chromosom X (47, XXY).
Objawy zespołu Klinefeltera
Obecność charakterystycznych objawów jest zależna od wieku chłopca oraz typu Zespołu Klinefeltera. Zdiagnozowanie zespołu następuje dość późno – zazwyczaj w okresie dojrzewania lub dopiero, gdy mężczyzna stara się bezskutecznie o potomstwo. Poniżej przedstawiono możliwe charakterystyczne objawy, zależne od etapu rozwoju chłopca.
Niemowlęta oraz dzieci
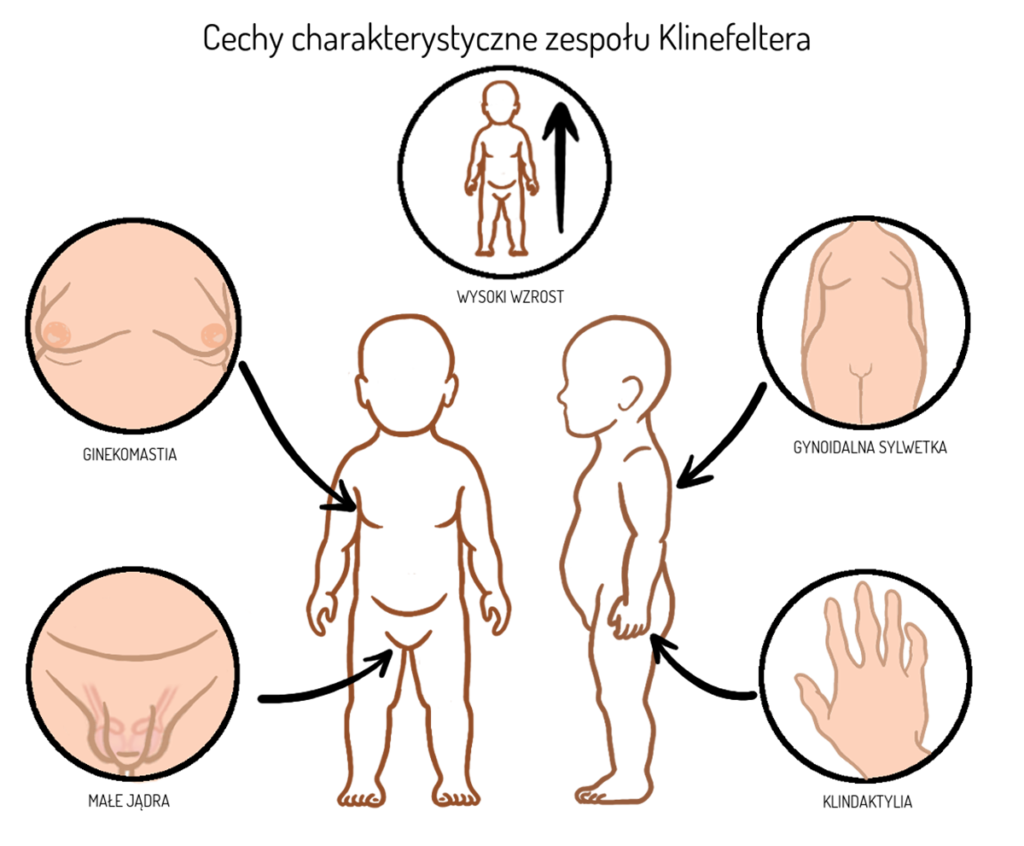
- początkowo prawidłowy wzrost, prawidłowa masa ciała oraz obwód czaszki. Wyższy wzrost (dłuższe kończyny) w porównaniu do grupy rówieśniczej obserwowany jest najczęściej między 5 a 8 rokiem życia,
- możliwe wystąpienie osłabienia siły mięśniowej (hipotonia)
- brak cech dysmorficznych twarzy (anomalii w budowie anatomicznej), możliwe utrzymanie się tzw. ‘chłopięcych’ rysów twarzy w okresie dorosłości,
- zaburzenia budowy zewnętrznych narządów płciowych: małe prącie (micropenis), niezstąpienie jąder do moszny,
- zespołowi nie towarzyszy specyficzna niepełnosprawność intelektualna, możliwe wystąpienie inteligencji w szerokim zakresie od poniżej przeciętnej do znacznie powyżej przeciętnej. Obliczono, że każdy dodatkowy chromosom X powoduje obniżenie poziomu inteligencji (IQ) o 15-16 punktów, a zatem w postaciach 48,XXXY oraz 49,XXXXY objawy są bardziej nasilone,
- deficyty mowy (trudności w formułowaniu wypowiedzi, nawet wyrażaniu pojedynczych słów) trudności w pisaniu, dysleksja, dysgrafia, problemy w rozumieniu złożonych gramatycznie zdań,
- deficyty uwagi, problemy z zapamiętywaniem,
- problemy w komunikowaniu się, wraz z trudnościami w słownym wyrażeniu emocji, prowadzącym do zaburzeń zachowania i postaw agresywnych a w efekcie także stanów depresyjnych.
Okres dojrzewania
- późniejsze niż u rówieśników wejście w okres dojrzewania,
- wyższy niż u rówieśników wzrost wraz z długimi kończynami utrzymuje się w okresie dojrzewania, ponadto obserwuje się krótszy tułów, szerszą miednicę oraz węższe barki,
- zmiana proporcji ciała tworząca tzw. sylwetkę eunochoidalną: możliwe nagromadzenie tkanki tłuszczowej w obrębie sutków (ginekomastia) z równoczesnym powiększeniem piersi, ponadto w okolicy podbrzusza, bioder oraz ud
- niska zdolność jąder do produkcji testosteronu, wynikająca z ich nieprawidłowego rozwoju, prowadzi generalnie do wyżej opisanych zaburzeń rozwoju typowo męskiej sylwetki oraz do nieprawidłowego rozwoju tzw. drugorzędowych cech płciowych (brak mutacji głosu, możliwy brak owłosienia ciała oraz twarzy), zaburzenia popędu i możliwe wahania nastroju.
Dorosłość
Zmiany obserwowane u dorosłych mężczyzn z Zespołem Klinefeltera są efektem zaburzeń w okresie dojrzewania, ponadto niedobór testosteronu powoduje zachwianie równowagi hormonalnej – wzrost produkcji gonadotropin przez przysadkę mózgową co niesie ze sobą istotne konsekwencje kliniczne,
- najczęściej brak plemników w nasieniu,
- możliwe podwyższenie masy ciała, rozwoju otyłości oraz chorób z nią związanych,
- wzrost ryzyka rozwoju nowotworu piersi, ryzyko względne prawie 200-krotnie wyższe niż u zdrowego mężczyzny, prawdopodobnie wynikające z zaburzonej równowagi między poziomem hormonów: estradiolu do testosteronu,
- wysokie ryzyko rozwoju chorób o podłożu autoimmunologicznym: tocznia rumieniowatego, zespołu Sjogrena, reumatoidalnego zapalenia stawów,
- powiązane powikłania endokrynologiczne, obejmujące cukrzycę, niedoczynność tarczycy (hipotyroidyzm), obniżoną czynność przytarczyc (hipoparatyroidyzm),
- możliwe wystąpienie zastoju żylnego, prowadzącego do rozwoju żylaków i owrzodzeń nóg.
Zespół Klinefeltera – rokowanie
Chłopcy z zespołem Klinefeltera, mimo borykania się z licznymi problemami będącymi efektem choroby, mogą uzyskać całkowitą niezależność od rodziców w okresie dorosłości. Wielu kończy edukację i funkcjonuje na relatywnie normalnym poziomie. Ponadto Zespół Klinefeltera nie wpływa na przewidywaną długość życia.
Jak w przypadku większości wrodzonych wad genetycznych, nie jest możliwe całkowite wyleczenie pacjenta. Natomiast kluczem do złagodzenia uciążliwych objawów jest jak najwcześniejsze zdiagnozowanie zespołu i odpowiednie prowadzenie pacjenta, obejmujące:
- terapię mowy oraz dodatkowy tryb indywidualnego nauczania w okresie dziecięco-szkolnym wraz z oceną psychoedukacyjną,
- fizjoterapia, zwłaszcza przy wystąpieniu hipotonii,
- terapia zajęciowa,
- terapia testosteronem, najlepiej rozpoczęta na początku okresu dojrzewania, prowadzi w pierwszej kolejności do uzyskania prawidłowego poziomu testosteronu w surowicy krwi, estradiolu, hormonu folikulotropowego (FSH) oraz hormonu luteinizującego (LH). Normalizacja proporcji hormonów sprzyja uzyskaniu prawidłowych proporcji ciała oraz drugorzędowych cech płciowych, natomiast nie leczy niepłodności, ginekomastii oraz małych jąder. Ponadto prowadzi do ogólnej poprawy zachowania oraz wydajności pracy, a także umożliwia poprawę jakości życia seksualnego. Terapia wpływa także długofalowo na zmniejszenie ryzyka chorób autoimmunologicznych, nowotworu piersi oraz osteoporozy.