Trisomia 13 - Zespół Patau
Spis treści
Zespół Patau
Zespół Patau (trisomia 13) jest to zespół złożonych wad wrodzonych spowodowany obecnością dodatkowego chromosomu 13 lub dodatkowego fragmentu chromosomu 13. Trisomia 13 jest trzecią co do częstości występowania trisomią autosomalną. Często wiąże się z bardzo ciężkimi wadami, które w 95% są letalne już na etapie ciąży. Całkowita częstość występowania tej aberracji chromosomowej jest szacowana na 1:5000 ciąż i na 1:10000 – 1:20000 żywo urodzonych dzieci.
Po raz pierwszy w literaturze medycznej opis przypadku trisomii 13 przedstawił duński lekarz Thomas Bartholin w 1957 roku. Trzy lata później, w 1960 roku amerykański genetyk Klaus Patau wraz ze swoim zespołem dokonali pierwszej analizy cytogenetycznej u pacjenta z trisomią 13. chromosomu, a wyniki ich badań zostały opublikowane w czasopiśmie Lancet. Stąd najbardziej popularna nazwa trisomii 13 – zespół Patau, chociaż wcześniej w literaturze funkcjonowała również nazwa zespół Bartholin-Patau.

Podłoże genetyczne zespołu Patau
Zespół Patau to aberracja chromosomowa 13. chromosomu. Jest związana z nieprawidłową (nadmiarową) ilością materiału genetycznego, która powoduje szereg bardzo poważnych wad anatomicznych i rozwojowych. Materiał genetyczny człowieka, który znajduje się w jądrze komórkowym zorganizowany jest w postaci chromosomów. Prawidłowo, u zdrowego człowieka liczba chromosomów jest stała i wynosi 46. Na tą liczbę składają się 23 pary chromosomów, w tym 22 to tzw. autosomy i 1 para allosomów (chromosomów płci). W zespole Patau ten prawidłowy układ jest zaburzony obecnością dodatkowego chromosomu 13 – całego lub jego fragmentu.
Z punktu widzenia analizy cytogenetycznej trisomia 13 (Zespół Patau) może mieć 3 różne podłoża:
- Całkowita trisomia 13 – występuje najczęściej i dotyczy ok. 80% przypadków zespołu Patau. Charakteryzuje się tym, że w każdej komórce organizmu osoby chorej znajduje się cały dodatkowy chromosom 13 (innymi słowy w każdej komórce są trzy, a nie jak powinno być prawidłowo – dwa chromosomy 13). Całkowita trisomia 13 jest wynikiem nieprawidłowego rozejścia się chromosomów homologicznych, czyli chromosomów tej samej pary w procesie gametogenezy. Jak wiemy, gamety (komórki jajowe i plemniki) powstają w wyniku podziałów mejotycznych (GAMETOGENEZA) ich komórek macierzystych. Mejoza to właściwie dwa następujące po sobie podziały, których celem jest redukcja ilości materiału genetycznego. Z komórki diploidalnej (2n), która zawiera 23 pary chromosomów (razem 46), powstają gamety, które są komórkami haploidalnymi (n), które posiadają 23 chromosomy. To wszystko jest potrzebne po to, aby po połączeniu się komórki jajowej z plemnikiem (zapłodnienie) powstała z powrotem prawidłowa komórka diploidalna z docelowym zestawem chromosomów, czyli 23 pary, która da początek nowemu organizmowi. Jeżeli w trakcie podziałów mejotycznych wystąpi błąd rozdziału chromosomów (tzw. nondysjunkcja), powstaną gamety z nieprawidłową ilością chromosomów. Nondysjunkcja chromosomów może mieć miejsce zarówno w trakcie pierwszego, jak i drugiego podziału mejotycznego. Całkowita trisomia 13, podobnie jak większość trisomii autosomalnych (za wyjątkiem trisomii 18) zdecydowanie częściej jest wynikiem błędnego rozdziału w trakcie pierwszego podziału mejotycznego. Chociaż wg danych literaturowych nondysjunkcja w drugim podziale mejotycznym zdarza się częściej niż w pozostałych trisomiach autosomów i może dotyczyć nawet do 37% przypadków. W 91% dodatkowy chromosom 13 jest chromosomem pochodzenia matczynego, a nieprawidłowy rozdział chromosomów w procesie oogenezy wiąże się z podwyższonym wiekiem matki.

- Częściowa trisomia 13 – może dotyczyć do 15% przypadków zespołu Patau. W częściowej trisomii 13 w komórkach znajduje się dodatkowy fragment chromosomu 13 (najczęściej jest to długie ramię tego chromosomu). Jest ona najczęściej wynikiem obecności zrównoważonej translokacji Robertsonowskiej u jednego z rodziców. Translokacja Robertsonowska to rodzaj translokacji (przemieszczenia się fragmentu chromosomu), który dotyczy chromosomów akrocentrycznych (czyli takich, gdzie centromer umieszczony jest bardzo blisko jednego z końców chromosomu, co powoduje, że jedno ramię chromosomu jest długie, a drugie bardzo krótkie). Charakterystyczne dla tego rodzaju translokacji jest to, że następuje fuzja centromerów i połączenie długich ramion dwóch chromosomów akrocentrycznych oraz najczęściej dochodzi do utraty krótkich ramion tych chromosomów. Ponieważ geny kodujące znajdują się na długich ramionach, to nosiciel takiej translokacji jest zdrowy (nie ma tutaj utraty, bądź nadmiaru materiału genetycznego). Natomiast inaczej może wyglądać sytuacja w przypadku potomstwa takiej osoby. Przemieszczenie się długiego ramienia chromosomu 13 i przyłączenie do innego chromosomu powoduje, że w procesie gametogenezy (GAMETOGENEZA) mogą powstać komórki płciowe z nieprawidłową ilością chromosomów: albo z dodatkowym (nadmiarowym) materiałem genetycznym – one po zapłodnieniu dadzą trisomiczną zygotę (zespół Patau) lub takie, w których będzie brakowało tego chromosomu i po zapłodnieniu dadzą monosomiczną zygotę (monosomia 13). Translokacja Robertsonowska chromosomu 13 to najczęściej połączenie długiego ramienia tego chromosomu z długim ramieniem chromosomu 14 (zdarza się 1 na 1300 osób), rzadziej z chromosomami 15, 22 lub drugim chromosomem 13 (1 na 50 000 osób), jeszcze rzadziej z chromosomem 21 (1 na 100 000 osób).

- Trisomia 13 typu mozaikowego – jest najrzadziej występującym typem trisomii 13, dotyczy ok. 5% przypadków. Najczęściej powstaje w wyniku błędu podczas podziałów mitotycznych (GAMETOGENEZA) komórek już po zapłodnieniu. Ponieważ ten błąd rozdziału chromosomów homologicznych w mitozie dotyczy jedynie części komórek, to charakterystyczne dla trisomii mozaikowej jest obecność dodatkowego chromosomu 13 tylko w części komórek organizmu. Mówiąc inaczej – w organizmie osoby z trisomią 13 typu mozaikowego występują 2 linie komórek pod względem kariotypu: jedna linia ma prawidłowy zestaw chromosomów (46 chromosomów) , a druga linia ma nieprawidłowy zestaw (47 chromosomów) z „nadprogramowym” chromosomem 13. Fenotyp osób z mozaikowym typem trisomii 13 różni się znacząco między osobami z tym typem zespołu Patau.

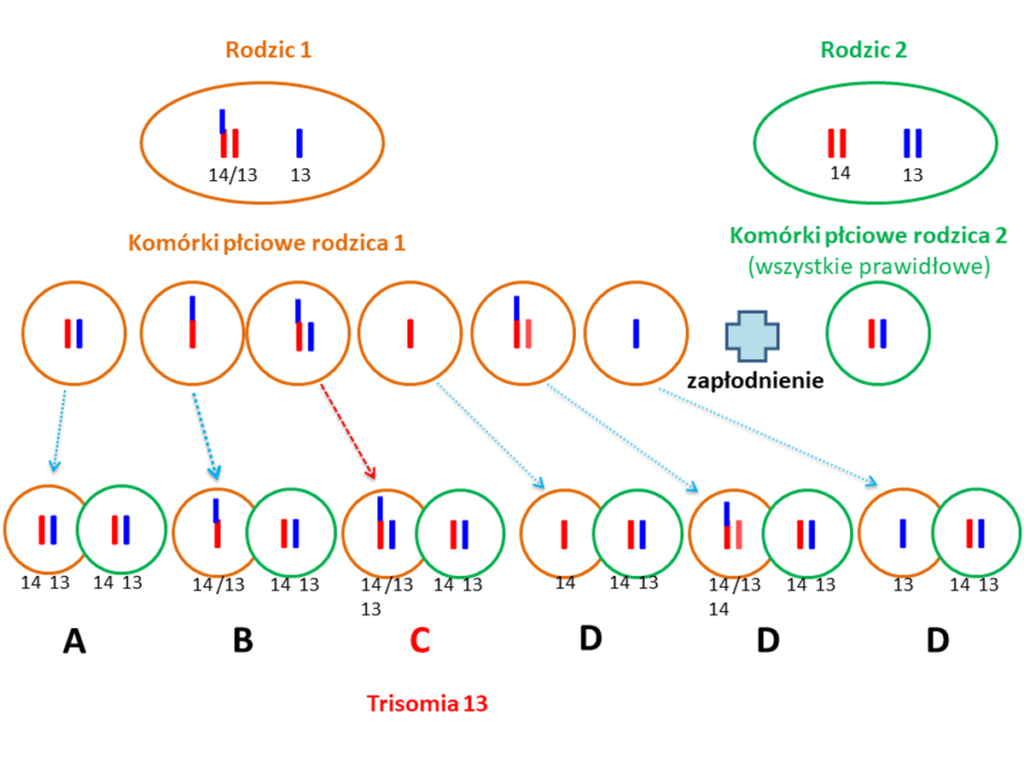
Czy rodzic, który posiada zrównoważoną translokację Robertsonowską zawsze przekaże ją swojemu potomstwu, a dziecko będzie miało zespół Patau?
Ryzyko urodzenia dziecka z zespołem Patau w sytuacji gdy rodzic jest nosicielem translokacji Robertsonowskiej chromosomu 13 szacuje się na ok. 1%. Dotyczy to sytuacji, gdy fragment chromosomu 13 jest przyłączony do innego chromosomu akrocentrycznego. W sytuacji gdy fragment chromosomu 13 jest połączony z homologicznym chromosomem 13 ryzyko urodzenia dziecka z trisomią 13 wynosi 100%.
W sytuacji pierwszej, czyli gdy u rodzica występuje zrównoważona translokacja Robertsonowska 13. Chromosomu (połączenie z innym chromosomem akrocentrycznym) istnieje kilka możliwości dla każdej z ciąż, w zależności od tego która z wytworzonych komórek rozrodczych ulegnie zapłodnieniu:
- (A) Dziecko może odziedziczyć prawidłowy zestaw 23 par chromosomów i nie będzie obciążone zespołem Patau.
- (B) Dziecko może odziedziczyć dokładnie taką samą zrównoważoną translokację Robertsonowską jaką ma rodzic. W takiej sytuacji zazwyczaj nie ma żadnych problemów zdrowotnych wynikających z takiej translokacji.
- (C) Dziecko może odziedziczyć translokację niezrównoważoną, czego efektem będzie trisomia 13.
- (D) Ciąża zakończy się poronieniem.
Poniższy schemat pokazuje możliwe opcje układu chromosomów 13. i 14. w komórkach płciowych rodziców, gdzie: rodzic 1 ma zrównoważoną translokację Robertsonowską (fragment chromosomu 13 jest połączony z chromosomem 14), a rodzic 2 ma prawidłowy układ chromosomów i możliwe opcje ich połączenia podczas zapłodnienia (literami A, B, C, D pokazane są schematycznie opisane powyżej opcje).
Chromosom 13
Chromosom 13 jest chromosomem akrocentrycznym, czyli jego centromer znajduje się bardzo blisko jednego końca chromosomu, co powoduje, że jedno ramię chromosomu 13 jest długie, a drugie bardzo krótkie. DNA tego chromosomu liczy ponad 114 milionów par zasad, co plasuje go mniej więcej w połowie, jeżeli chodzi o długość chromosomów pod względem ilości nukleotydów. Pomimo to, jest chromosomem o najniższej gęstości genowej wśród autosomów.
Chromosom 13 ma 6,5 genu na 1 mln par zasad (Mb), a dla porównania gęstość genowa chromosomu 22, którego DNA ma długość jedynie 50 milionów par zasad to 16,3 genów na 1 Mb. Aktualnie znana liczba genów kodujących chromosomu 13 to 322 geny (Esemble genome browser release, 2021). Badania wskazują, że główną rolę w trisomii 13 pełni długie ramię tego chromosomu, gdzie zlokalizowane są geny kodujące.

Fenotyp trisomii 13 jest bardzo zróżnicowany. W obrazie klinicznym całkowitej trisomii chromosomu 13 najczęściej obserwujemy ciężkie malformacje ośrodkowego układu nerwowego jak holoprosencefalia, upośledzenie umysłowe wysokiego stopnia, zaburzenia wzrostu, rozwoju motorycznego, rozszczep wargi i podniebienia, małoocze lub bezocze oraz polidaktylię, wrodzone wady serca i nerek. Szerzej obraz kliniczny tej aberracji chromosomowej opisujemy poniżej w części „Objawy trisomii 13”. Nie jest do końca jasne, które „potrojone” regiony chromosomu 13 są odpowiedzialne za poszczególne, wyżej wymienione nieprawidłowości rozwojowe w całkowitej trisomii 13. Dostępne analizy poszczególnych przypadków zespołu Patau wskazują na to, że za większość objawów trisomii 13 odpowiada nie jeden, ale kilka regionów chromosomu. Stąd trudniej jest precyzyjnie określić i dopasować geny, czy też grupy genów bądź regiony chromosomu 13 zaangażowane w powstawanie konkretnych wad rozwojowych. Pomocne w tym obszarze mogą być badania pacjentów z częściową trisomią chromosomu 13, które w pewnym stopniu przybliżają nas do odpowiedzi na to pytanie. Nadal jednak wiele kwestii wymaga dalszych testów i analiz. Badania cytogenetyczne chorych z częściową trisomią 13 pozwoliły na wyłonienie trzech dość szerokich (w kontekście fenotypowym) grup pacjentów. W tym celu przeanalizowano dane 190 chorych.
Biorąc pod uwagę fenotyp zespołu Patau i region długiego ramienia chromosomu 13 (13q), który u danego pacjenta z częściową trisomią 13 był potrojony wyodrębniono następujące grupy:
- Trisomia części centralnej 13q (długiego ramienia chromosomu 13) – obejmuje segment 13q2 (13q21-22). W tym regionie znajduje się stosunkowo mało genów, w porównaniu do części dystalnej i proksymalnej długiego ramienia, stąd osoby z częściową trisomią 13. chromosomu, która jest wynikiem potrojenia materiału genetycznego tego regionu mają najmniej poważnych wad rozwojowych. Wśród przeanalizowanych 40 przypadków były to: mikrocefalia (2 osoby), wrodzone wady serca (4 osoby), wiotkość stawów (6 osób) oraz pojedyncze przypadki takich wad jak atrezja przełyku, wrodzone zwężenie nozdrzy tylnych, torbielowatość nerek, zaćma. Wśród zaburzeń funkcjonalnych u 4 pacjentów występowały drgawki, u kolejnych czterech ubytek słuchu, a u dwóch osób autyzm. Uważa się, że trisomia części centralnej 13q jest najłagodniejsza i najczęściej nie daje charakterystycznych objawów zespołu Patau.
- Trisomia części dystalnej 13q (długiego ramienia chromosomu 13) – obejmuje odcinek 13q21qter lub 13q22qter (od segmentu 13q.21 lub 13q.22 do końca ramienia długiego chromosomu 13). W literaturze najwięcej przypadków częściowej trisomii 13 dotyczy właśnie tej części 13q. Przeanalizowano grupę 115 pacjentów i ogólnie, w tej grupie widzimy większość objawów całkowitej trisomii 13, chociaż częstotliwość ich występowania jest dużo niższa:
– Praktycznie wszyscy pacjenci charakteryzowali się zmniejszonym obwodem głowy, ale holoprosencefalia, czyli najczęstsza i najpoważniejsza wada mózgu był obecna jedynie u 6% pacjentów, podczas gdy w całkowitej trisomii 13 jest ona obecna u 50% osób. Prawdopodobnie wynika to z tego, że holoprosencefalia jest powiązana z przynajmniej 2 regionami chromosomu 13 i genu zlokalizowane w tych segmentach muszą współdziałać, aby wystąpiła ta wada. Szukano również powiązania genu ZIC2 z holoprosencefalią w triosmii 13. Gen ZIC2 położony jest w regionie 13q32.2 i delecja tego genu jest głównym czynnikiem powstania tej wady w innej chorobie genetycznej – zespole delecji 13q. Jednak nie znaleziono potwierdzenia, jakoby trisomia tego genu była związana z holoprosencefalią. Za to u 15% pacjentów występowała trigonocefalia, a regionem krytycznym związanym z występowaniem tej wady jest region 13q31.3 – 13q33.1. Z kolei region znajdujący się w jego środku 13q32q33 odpowiedzialny jest za niedorozwój lub agenezję ciała modzelowatego
– U prawie 40% pacjentów występowały wady oczu (mikroftalmia lub anoftalmia, coloboma. Za region krytyczny dla tych nieprawidłowości uważany jest region 13q32.3 – 13q33.1.
– Rozszczepienie podniebienia i/lub wargi rozpoznano u 13% pacjentów. Z tym, że pacjenci z trisomią 13q22qter (lub większą) mieli jedynie rozszczep wargi, a z kolei pacjenci z trisomią bardziej dystalnych segmentów (13q31qter) mieli jedynie rozszczep podniebienia, co wskazuje na to, że istnieją co najmniej 2 grup genów na długim ramieniu chromosomu 13 zaangażowane w powstawanie tej wady.
– Polidaktylia pozaosiowa (dodatkowy palec lub palce po stronie małego palca, czyli po stronie kości łokciowej u ręki lub kości strzałkowej u nogi) była obecna u 40% pacjentów. Najbardziej prawdopodobna lokalizacja genów odpowiedzialnych za jej występowanie to region 13q31.
– Wrodzone wady serca, które dotyczą ok. 80% dzieci urodzonych z całkowitą trisomią 13, występują również u pacjentów z częściową trisomią 13 wynikającą z trisomii dystalnej części długiego ramienia chromosomu 13., ale są rzadsze (ok. 22%), i najczęściej można je skutecznie leczyć bądź ich występowanie nie ma poważnych skutków w rozumieniu zagrożenia życia (np. dekstrokardia, ubytek przegrody międzykomorowej, przetrwały przewód tętniczy, zwężenie aorty). Najprawdopodobniej regionów, których trisomia wiąże się z wrodzonymi wadami serca jest co najmniej kilka.
– Zdecydowanie rzadziej u pacjentów z trisomią dystalnej części 13q występują wady nerek (10%) i wady przewodu pokarmowego (5%), natomiast dość powszechne w tej grupie są napady drgawek oraz niedosłuch.Proksymalna trisomia 13q (długiego ramienia chromosomu 13) – obejmuje region od centromeru do 13q21. Ta grupa jest najbardziej różnorodna zarówno fenotypowo, jak i pod względem cytogenetycznym (rozmiaru regionu, który jest potrojony, nie wszyscy pacjenci w tej grupie mają dokładnie potrojony cały odcinek od centromeru do 13q21). W opublikowanych analizach (25 przypadków) większość chorych charakteryzował opóźniony rozwój psychomotoryczny, wady układu pokarmowego (malrotacja jelit) i nerek (wrodzone poszerzenia układu kielichowo-miedniczkowego, podwojenie moczowodów, wielotorbielowatość nerek), wady oczu (mikroftalmia, coloboma, zaćma, jaskra). Wrodzone wady serca występowały u 25% pacjentów tej grupy (zwężenie cieśni aorty, ubytek przegrody przedsionkowo-komorowej – AVSD, tetralogia Fallota). Wśród chorych z proksymalną trisomią 13q były pojedyncze przypadki nieprawidłowości związanych z rozwojem układu nerwowego (holoprosencefalia , hydrocefalia, mikrocefalia, niedorozwój ciała modzelowatego, rozszczep wargi, rozszczep podniebienia), wad narządu słuchu i polidaktylii. Jak widzimy, obraz kliniczny w tej grupie nie jest jednorodny, część nieprawidłowości rozwojowych występuje częściej, inne dotyczą 1-5% chorych z trisomią proksymalnego regionu długiego ramienia chromosomu 13. Wszystkie te objawy są również typowe dla całkowitej trisomii 13 oraz są bardzo częste w przypadku częściowej trisomii dystalnego regionu długiego ramienia chromosomu 13. Można więc wysnuć wniosek, że na chromosomie 13 znajduje się kilka segmentów rozmieszczonych w różnych częściach chromosomu, a odpowiedzialnych za ten sam rodzaj nieprawidłowości (w momencie gdy są potrojone).

Czynniki ryzyka urodzenia dziecka z zespołem Patau
- ryzyko urodzenia dziecka posiadającego dodatkowy chromosom 13 pary wzrasta wraz z wiekiem matki; pomimo, że ten wzrost ryzyka jest znaczący, to jest on mniej gwałtowny niż w przypadku zespołu Downa czy zespołu Edwardsa (np. u kobiet w 35. roku życia ryzyko urodzenia dziecka z zespołem Patau jest dwukrotnie wyższe niż u kobiet w 30 roku życia, kolejno w 40 i 45 roku życia w porównaniu do 30 roku życia to ryzyko wzrasta siedmiokrotnie i piętnastokrotnie, podczas gdy ryzyko urodzenia dziecka z zespołem Edwardsa wzrasta odpowiednio dziesięcio- i trzydziestotrzykrotnie)
- obecność zrównoważonej translokacji Robertsonowskiej z zaangażowaniem chromosomu 13 u jednego z rodziców
- ryzyko ponownego urodzenia dziecka z zespołem Patau wynosi ok. 1%
Objawy trisomii 13 (Zespół Patau)
Dzieci z zespołem Patau rodzą się z niską masą urodzeniową i niskim napięciem mięśniowym, szeregiem wad anatomicznych twarzoczaszki, kończyn oraz wadami układowymi (wady serca, układu nerwowego, narządów zmysłów, układu moczowo-płciowego, układu pokarmowego) powodującymi szereg problemów zdrowotnych. Do najczęściej występujących wad należą wady oczu (mikroftalmia, anoftalmia, hipoteloryzm), rozszczep wargi lub podniebienia, polidaktylia, wady serca i holoprosencefalia. Trisomia 13 wiąże się również z wysokim stopniem upośledzenia psychoruchowego i umysłowego.
Zespół objawów zespołu Patau różni się między chorymi dziećmi i nie wszystkie wymienione poniżej objawy występują na raz u jednej osoby. Występowanie poszczególnych zaburzeń i stopień ich nasilenia zależy m.in. od tego jak duża ilość materiału genetycznego 13. chromosomu występuje w trzeciej kopii i której części chromosomu ona dotyczy. Niemniej jednak zespół Patau uznawany jest za wadę letalną, a liczne wady stanowią bardzo duże obciążenie dla zdrowia dziecka z tym zespołem.

Wady i schorzenia towarzyszące zespołowi Patau
Wady czaszki i twarzy w trisomii 13
- mikroftalmia (małoocze) – gałka oczna w okresie życia płodowego nie osiąga prawidłowych rozmiarów, co powoduje, że ostatecznie jest mniejsza niż powinna być w określonym wieku dziecka; mikroftalmia może dotyczyć jednego oka lub obu oczu (dotyczy 50-70% chorych)
- anoftalmia (bezocze) – gałka oczna nie rozwinęła się w życiu płodowym, wyróżnić możemy tzw. prawdziwy brak gałki ocznej (nie ma nerwu wzrokowego ani zawiązka gałki ocznej) lub rzekomy brak gałki ocznej (nerw wzrokowy jest rozwinięty i w worku spojówkowym znajduje się zawiązek niewykształconej gałki ocznej, która jest niezdolna do widzenia)
- trigonocefalia – przedwczesne zarośnięcie oraz kostnienie szwu czołowego, które prowadzi do trójkątnego zniekształcenia czoła
- hipoteloryzm – mała odległość pomiędzy gałkami ocznymi
- cyklopia – połączenie oczodołów i wykształcenie jednej gałki ocznej
- rozszczep podniebienia lub wargi (60-70% chorych)
- atrezja nozdrzy tylnych – wada wrodzona charakteryzująca się niedrożnością jednego (jednostronna) lub obydwu (obustronna) otworów wewnętrznych nosa, spowodowana obecnością kostnej lub błoniastej przegrody pomiędzy nosem a jamą nosowo-gardłową, może być przyczyną ostrej niewydolności oddechowej
- bulwiasty nos (wydatny grzbiet nosa)
- płaska potylica
- mikrognatyzm (mikrogenia, hipoplazja) żuchwy – niedorozwój żuchwy
- nisko osadzone małżowiny uszne
- mikrocefalia (małogłowie) – patologicznie małe wymiary czaszki , a co za tym idzie mała masa mózgu
- liczne naczyniaki włośniczkowe na skórze, zwłaszcza w okolicy czoła
- ubytek skóry na głowie

Wady układu ruchu w zespole Patau
- zrośnięcie palców lub obecność palców dodatkowych (syndaktylia, polidaktylia) – dotyczy ok. 75% osób z trisomią 13
- niedorozwój paznokci
- zagięcie palucha stopy
- wrodzona stopa płaska (stopa suszkowata, stopa typu rocker bottom) – sklepienie podłużne stopy jest odwrócone, a pięta i przedstopie są ustawione wysoko
- zgięcie podeszwowe bliższej części stopy z jednoczesnym zgięciem grzbietowym dalszej części stopy (przodostopia)
- pojedyncza, gruba bruzda zgięciowa (tzw. małpia bruzda) – bruzda biegnąca w poprzek wewnętrznej części dłoni w miejscu gdzie większość ludzi ma dwie płytsze bruzdy
- rzadziej ektrodaktylia (tzw. szczypce homara) – całkowity lub częściowy brak palców stóp i/lub dłoni
- przykurcze stawów
- skolioza, kifoza
Wady ściany jamy brzusznej w trisomii 13
- przepuklina pępowinowa
- przepuklina przeponowa
Wady wrodzone serca w trisomii 13
Występują u ok. 80% urodzonych dzieci z zespołem Patau.
- ubytek w przegrodzie przedsionkowo-komorowej, międzykomorowej lub międzyprzedsionkowej
- przetrwały przewód tętniczy
- dekstrokardia – prawostronne położenie serca
- tetralogia Fallota (wada serca, która obejmuje cztery nieprawidłowości anatomiczne: ubytek w przegrodzie międzykomorowej, zwężenie zastawki pnia płucnego, z którym może współistnieć zwężenie pnia płucnego czy też odejścia prawej lub lewej tętnicy płucnej, dekstropozycja aorty, czyli przemieszczenie aorty na prawo nad przegrodę międzykomorową oraz przerost prawej komory serca

Wady układu nerwowego w zespole Patau
- holoprosencefalia -wada mózgowia wynikającą z zaburzonego podziału przodomózgowia na dwie odrębne półkule, gdzie komory boczne są zastąpione przez jedną wspólną komorę; często uszkodzone są również środkowe struktury przodomózgowia takie jak wzgórze, podwzgórze, przysadka mózgowa, gałki oczne i drogi wzrokowe, często nieprawidłowej budowie ośrodkowego układu nerwowego towarzyszą poważne zaburzenia rozwoju linii środkowej twarzy
- agenezja ciała modzelowatego, czyli spoidła wielkiego mózgu – brak lub niepełne wykształcenie ciała modzelowatego, czyli struktury łączącej obie półkule mózgowe odpowiadającej za przekazywanie informacji pomiędzy nimi, spoidło mózgu odgrywa istotną rolę w czynności poznawczej mózgu
- arhincefalia – niepełne wykształcenie płatów czołowych mózgu
- hipoplazja móżdżku – niepełne wykształceniu móżdżku
- brak opuszek węchowych
- dysrafie układu nerwowego – wady wynikające z zaburzeń w tworzeniu i zamykaniu cewy nerwowej na etapie życia płodowego (np. anencefalia, czyli bezmózgowie częściowe lub całkowite, przepuklina mózgowa, oponowa, oponowo-rdzeniowa, zespół Arnolda-Chiariego, jamistość rdzenia kręgowego)
- zespół Dandy’ego-Walkera (malformacja Dandy’ego-Walkera) – zespół wad wrodzonych tyłomózgowia
Wrodzone wady i schorzenia narządu wzroku oraz słuchu u chorych z trisomią 13
- coloboma, tzw. rozszczep tęczówki i ciałka rzęskowego – wada charakteryzująca ubytkiem, rozszczepem w strukturze tęczówki oka oraz ciałka rzęskowego (część gałki ocznej, która łączy tęczówkę z naczyniówką), występuje u 25-30% osób z zespołem Patau
- wady siatkówki oka
- malformacje ucha
- niedosłuch czuciowo – nerwowy – ubytek słuchu, który wynika z uszkodzenia lub braku komórek słuchowych ślimaka i/lub struktur je otaczających lub jest związany z zaburzeniami jakości przewodzenia bodźca w nerwie słuchowym oraz jego odbioru i interpretacji w mózgu
- głuchota
Wady układu moczowo-płciowego w trisomii 13
- wielotorbielowatość nerek (65-70% osób z zespołem Patau)
- spodziectwo (hypospadiasis) – wada rozwojowa układu moczowo-płciowego u chłopców, która charakteryzuje się tym, że ujście zewnętrzne cewki moczowej często jest zwężone i znajduje się w nieprawidłowym położeniu – na brzusznej powierzchni prącia, zwężenie znacznego stopnia może być przyczyną poważnych zmian w obrębie górnych dróg moczowych (np. odpływ pęcherzowo-moczowodowy, czyli cofanie się moczu z pęcherza moczowego do moczowodów i układu kielichowo-miedniczkowego nerki)
- wierzchniactwo (epispadiasis) – wrodzona wada strukturalna dróg moczowych, która może występować zarówno u dziewczynek, jak i chłopców jako wada samodzielna lub występująca w zespole (częściej) z wynicowanym pęcherzem moczowym; u chłopców charakteryzuje się rozszczepieniem cewki moczowej, która jest nieprawidłowo położona – na górnej powierzchni prącia, grzbietowe rozszczepienie powoduje, że narząd ten nabiera wygląd przypominający rynienkę; u dziewczynek występuje rozdzielenie (rozsunięcie) ciał jamistych łechtaczki, rozszczep cewki, zaburzenie budowy szyi pęcherza, a czasem również rozejście spojenia łonowego; wierzchniactwo może prowadzić do nietrzymania moczu
- wodonercze
- macica dwurożna
- mikropenis
- wnętrostwo – brak jednego lub obu jąder w mosznie z jednoczesną obecnością ich w jamie brzusznej lub kanale pachwinowym

Wady układu pokarmowego w trisomii 13
- atrezja przełyku
- zwężenie lub atrezja jelita grubego lub odbytnicy
- choroba Hirschsprunga – wrodzone nieprawidłowe unerwienia jelita, które powoduje, że dotknięty chorobą odcinek jelita jest w stanie ciągłego skurczu, co powoduje jego niedrożność
- przetoki jelitowe
- malrotacja jelit (niedokonany zwrot jelit) – grupa zaburzeń rozwojowych, która związana jest z nieprawidłowym przebiegiem skrętu jelita który może prowadzić do niedrożności jelit
U dzieci z zespołem Patau obserwuje się również częste nawracające infekcje, ciężkie upośledzenie umysłowe, poważne opóźnienia w rozwoju psychomotorycznym, występowanie drgawek i bezdechów sennych.
Nowotwory a zespół Patau
U osób z trisomią 13 chorych na ostrą białaczkę szpikową stwierdzono zwiększoną ekspresję genu FLT3, który jest zlokalizowany na chromosomie 13. Gen ten koduje białko, tzw. receptorową kinazę tyrozynową (białko FLT3). Receptor dla tego białka występuje w komórkach macierzystych szpiku. W prawidłowych warunkach fizjologicznych połączenie tego receptora z białkiem FLT3 powoduje pobudzenie sygnałów wewnątrzkomórkowych regulujących proliferację (namnażanie) i różnicowanie komórek. W komórkach białaczkowych często obserwuje się stałą, niezależną od białka FLT3 aktywację tego receptora, co prowadzi do niekontrolowanej proliferacji komórek, a więc do onkogenezy. Z kolei ekspresja innego genu chromosomu 13 – genu SPRY2 jest znacząco obniżona u osób z zespołem Patau. Gen ten znany jest jako tzw. the tumor suppressor gene, czyli gen „hamujący nowotworzenie” (m.in. dlatego, że białko kodowane przez ten gen działa na zasadzie ujemnego – hamującego sprzężenia zwrotnego dla receptora FLT3). Niska ekspresja tego genu powoduje niski poziom białka SPRY2, a więc nie dochodzi do hamowania receptora FLT3, a co za tym idzie nadal następuje niekontrolowane namnażanie się komórek w szpiku.
Ponadto, badania wskazują, że trisomia 13 wiąże się z mutacjami i zwiększoną ekspresją genu RUNX1, który jest położony na chromosomie 21. Prawidłowa funkcja genu RUNX1 obejmuje m.in. kontrolę procesu hematopoezy w rozwoju zarodkowym. Gen ten bierze udział w powstawaniu oraz dojrzewaniu krwiotwórczych komórek progenitorowych oraz komórek macierzystych. Gen RUNX1 kontroluje również ekspresję innych genów, m.in. genów zaangażowanych w różnicowanie hematopoetyczne oraz regulację cyklu komórkowego. Zwiększona ekspresja genu RUNX1 może być onkogenna i występuje w ostrej białaczce szpikowej (AML, ang. acute myeloid leukemia), ostrej białaczce limfoblastycznej (ALL, ang. acute lymphoblastic leukemia) oraz w różnych nowotworach nabłonkowych (m.in. rak skóry, rak endometrioidalny). Mutacje genu RUNX1 są bardzo częste w trisomii 13 (75-80%). Nie jest jeszcze jasne i wymaga to dalszych badań, jakie jest powiązanie pomiędzy ekspresją genu RUNX1, a genami FLT3 czy SPYR2.
Dane te wskazują na to, że istnieje ścisły związek pomiędzy mutacjami i ekspresją genów charakterystycznych dla białaczki szpikowej u osób z zespołem Patau. Przebadane grupy osób z trisomią 13 chorych na AML były bardzo homogenne genetycznie w kontekście genów związanych z białaczką, co może być w przyszłości podłożem do badań nad nowymi strategiami terapeutycznymi m.in. białaczki szpikowej. Kierunek ten wydaje się być szczególnie interesujący, gdyż wymienione mutacje występują również u części osób z AML, ale bez aneuploidii i wiążą się ze złymi rokowaniami. Niemniej jednak, ponieważ grupa osób z zespołem Patau jest niewielka i ich okres życia jest raczej krótki, badania te wymagają czasu. Zrozumienia wymaga również wpływ wymienionych genów na siebie.
Diagnostyka zespołu Patau
Diagnostyka zespołu Patau, podobnie jak w przypadku trisomii 21 i 18 rozpoczyna się już na etapie ciąży. W pierwszym trymestrze ciąży przeprowadzane są badania przesiewowe, dzięki którym można ocenić ryzyko wystąpienia trisomii 13 u płodu.
Pomiędzy 11. a 14. tygodniem ciąży każda kobieta ciężarna powinna mieć zalecone USG prenatalne (genetyczne) pierwszego trymestru (USG PRENATALNE PIERWSZEGO TRYMESTRU). Podczas badania poza oceną budowy płodu, analizuje się tzw. markery wad genetycznych (główny marker – przezierność karkowa – NT, oraz ewentualnie dodatkowe markery, jakimi są: kość nosowa – NB, przepływ krwi przez przewód żylny – DV przepływ krwi przez zastawkę trójdzielną – TR).

Drugim zalecanym badaniem, jest test podwójny (TEST PAPP-A), czyli badanie biochemiczne, które mierzy stężenie dwóch substancji: białka ciążowego A (tzw. białka PAPP-A) oraz wolnej podjednostki beta-hCG, czyli hormonu wydzielanego przez łożysko. Zgodnie z rekomendacjami Polskiego Towarzystwa Ginekologicznego, optymalny czas do pobrania krwi od kobiety ciężarnej, aby wykonać do badanie to 10. – 11. tydzień ciąży).
Zestawienie i ocena wyników tych dwóch badań przesiewowych (markery wad genetycznych w USG oraz poziom białka PAPP-A i wolnej podjednostki beta-hCG) przy użyciu specjalnych algorytmów (tzw. test zintegrowany) pozwala ocenić ryzyko wystąpienia trisomii 13 u płodu.
Jeżeli potrzebne jest więcej informacji, od 9.-10. tygodnia ciąży można również zbadać krew kobiety w ciąży poprzez test typu NIPT (CO TO JEST NIPT) (nieinwazyjny test prenatalny, jak np. Harmony ,Nace, Neobona, Nifty, Nifty Pro, Panorama, Sanco, Veragene, Veracity), który bada wolne płodowe DNA (cffDNA, ang. cell free fetal DNA). Jednak, co ważne – wynik takiego testu NIPT również nie daje nam pewności, a określa ryzyko wystąpienia trisomii.
Aby ostatecznie potwierdzić lub wykluczyć zespół Patau, trzeba ocenić kariotyp płodu w badaniu cytogenetycznym (ilościowa i jakościowa ocena zestawu chromosomów). Taką możliwość dają inwazyjne badania prenatalne, m.in. amniopunkcja (AMINOPUNKCJA), która jest zalecana jako badanie diagnostyczne, w sytuacji gdy ryzyko trisomii wyliczone w teście zintegrowanym wynosi > 1:300. Ponieważ badania te należą do grupy badań inwazyjnych, ich wykonanie wiąże się z niewielkim ryzykiem powikłań ciąży, dlatego to ostatecznie rodzice po rozmowie z lekarzem podejmują decyzję czy takie badanie chcą wykonać. Ocenę kariotypu można również wykonać po urodzeniu.
Kariotyp człowieka podawany jest w formie zapisu:
- kariotyp prawidłowy – zapis dla odpowiednio chłopca i dziewczynki to: 46XY lub 46XX
- kariotyp zespołu Patau – zapis dla odpowiednio chłopca i dziewczynki to: 47XY,+13 lub 47XX,+13, co oznacza, że zestaw chromosomów posiada dodatkowy chromosom (zamiast 46, jest ich 47) z zaznaczeniem, który z chromosomów jest nadmiarowy (+13 – czyli chromosom 13)
Po urodzeniu dziecko z zespołem Patau wymaga rozległej i wielokierunkowej diagnostyki oraz opieki medycznej. Jej zakres zależy od stanu zdrowia i potrzeb zdrowotnych dziecka.
Leczenie trisomii 13
Trisomia 13. chromosomu to zespół wad genetycznych, którego nie potrafimy skutecznie leczyć. Stosowane leczenie ma głównie charakter objawowy i paliatywny mający na celu poprawę komfortu życia dziecka. Niektóre wady (np. wady serca, układu pokarmowego, wady układu kostno-stawowego) można poddać zabiegom chirurgicznym Wszystko jest uzależnione od stanu zdrowia chorego. Ze względu na złożoność i szeroki zakres wad i schorzeń dziecko z zespołem Patau wymaga wielospecjalistycznej opieki, dostosowanej do jego potrzeb. Począwszy od lekarza POZ, przez innych specjalistów jak np. kardiolog, nefrolog, neurolog, chirurg, okulista czy fizjoterapeuta. Z uwagi na obecność ciężkich wad wrodzonych prowadzących do licznych powikłań dzieci z zespołem Patau wymagają stałej opieki. Ich rozwój fizyczny, psychomotoryczny i intelektualny jest uzależniony od stopnia zaawansowania poszczególnych wad, ale najczęściej jest on upośledzony w stopniu ciężkim.
Rokowanie w zespole Patau
Zespół Patau jest jednym z najcięższych zespołów wad genetycznych i jest zaliczany do wad letalnych. W około 90% przypadków dochodzi do samoistnego poronienia lub martwego urodzenia. Rokowania dla dzieci żywo urodzonych są złe. Średnia przeżycia po urodzeniu to 7-10 dni, a 86-91% dzieci nie przeżyje dłużej niż rok. Najnowsze dane pokazują, że tylko 9,7% dzieci z trisomią 13 osiągnie 5. rok życia. Znane jest kilka pojedynczych przypadków dłuższego okresu życia dzieci z zespołem Patau – najdłuższe udokumentowane opisy dotyczą 19-letniej pacjentki z całkowitą trisomią 13 i 38-letniego pacjenta z trisomią 13 typu mozaikowego.
Bibliografia
- Noriega MA, Siddik AB. Trisomy 13. 2021 Jul 7. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan–. PMID: 32644517.
- Williams GM, Brady R. Patau Syndrome. 2021 Jul 2. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan–. PMID: 30855930.
- Cuckle H, Morris J. Maternal age in the epidemiology of common autosomal trisomies. Prenat Diagn. 2021 Apr;41(5):573-583. doi: 10.1002/pd.5840. Epub 2020 Oct 19. PMID: 33078428.
- Kevin L Howe, Premanand Achuthan, James Allen, Jamie Allen, Jorge Alvarez-Jarreta, M Ridwan Amode, Irina M Armean, Andrey G Azov, Ruth Bennett, Jyothish Bhai, Konstantinos Billis, Sanjay Boddu, Mehrnaz Charkhchi, Carla Cummins, Luca Da Rin Fioretto, Claire Davidson, Kamalkumar Dodiya, Bilal El Houdaigui, Reham Fatima, Astrid Gall, Carlos Garcia Giron, Tiago Grego, Cristina Guijarro-Clarke, Leanne Haggerty, Anmol Hemrom, Thibaut Hourlier, Osagie G Izuogu, Thomas Juettemann, Vinay Kaikala, Mike Kay, Ilias Lavidas, Tuan Le, Diana Lemos, Jose Gonzalez Martinez, José Carlos Marugán, Thomas Maurel, Aoife C McMahon, Shamika Mohanan, Benjamin Moore, Matthieu Muffato, Denye N Oheh, Dimitrios Paraschas, Anne Parker, Andrew Parton, Irina Prosovetskaia, Manoj P Sakthivel, Ahamed I Abdul Salam, Bianca M Schmitt, Helen Schuilenburg, Dan Sheppard, Emily Steed, Michal Szpak, Marek Szuba, Kieron Taylor, Anja Thormann, Glen Threadgold, Brandon Walts, Andrea Winterbottom, Marc Chakiachvili, Ameya Chaubal, Nishadi De Silva, Bethany Flint, Adam Frankish, Sarah E Hunt, Garth R IIsley, Nick Langridge, Jane E Loveland, Fergal J Martin, Jonathan M Mudge, Joanella Morales, Emily Perry, Magali Ruffier, John Tate, David Thybert, Stephen J Trevanion, Fiona Cunningham, Andrew D Yates, Daniel R Zerbino, Paul Flicek, Ensembl 2021, Nucleic Acids Research, Volume 49, Issue D1, 8 January 2021, Pages D884–D891.
- Outtaleb FZ, Errahli R, Imelloul N, Jabrane G, Serbati N, Dehbi H. La trisomie 18 ou syndrome d’Edwards en post-natal: étude descriptive au Centre Hospitalier Universitaire de Casablanca et revue de littérature [Trisomy 18 or postnatal Edward´s syndrome: descriptive study conducted at the University Hospital Center of Casablanca and literature review]. Pan Afr Med J. 2020 Dec 3;37:309. French. doi: 10.11 Meyer RE, Liu G, Gilboa SM, Ethen MK, Aylsworth AS, Powell CM, Flood TJ, Mai CT, Wang Y, Canfield MA; National Birth Defects Prevention Network. Survival of children with trisomy 13 and trisomy 18: A multi-state population-based study. Am J Med Genet A. 2016 Apr;170A(4):825-37. doi: 10.1002/ajmg.a.37495. Epub 2015 Dec 10. PMID: 26663415; PMCID: PMC4898882.604/pamj.2020.37.309.26205. PMID: 33654528; PMCID: PMC7896527.
- Goel N, Morris JK, Tucker D, de Walle HEK, Bakker MK, Kancherla V, Marengo L, Canfield MA, Kallen K, Lelong N, Camelo JL, Stallings EB, Jones AM, Nance A, Huynh MP, Martínez-Fernández ML, Sipek A, Pierini A, Nembhard WN, Goetz D, Rissmann A, Groisman B, Luna-Muñoz L, Szabova E, Lapchenko S, Zarante I, Hurtado-Villa P, Martinez LE, Tagliabue G, Landau D, Gatt M, Dastgiri S, Morgan M. Trisomy 13 and 18-Prevalence and mortality-A multi-registry population based analysis. Am J Med Genet A. 2019 Dec;179(12):2382-2392. doi: 10.1002/ajmg.a.61365. Epub 2019 Sep 30. PMID: 31566869; PMCID: PMC6848757..
- Goff RD, Soares BP. Neuroradiological findings of trisomy 13 in a rare long-term survivor. Neuroradiol J. 2018 Aug;31(4):412-414. doi: 10.1177/1971400916689575. Epub 2017 Feb 14. PMID: 28195512; PMCID: PMC6111439.
- Peterson JK, Kochilas LK, Catton KG, Moller JH, Setty SP. Long-Term Outcomes of Children With Trisomy 13 and 18 After Congenital Heart Disease Interventions. Ann Thorac Surg. 2017 Jun;103(6):1941-1949. doi: 10.1016/j.athoracsur.2017.02.068. Epub 2017 Apr 26. PMID: 28456396; PMCID: PMC5526352.
- Imataka G, Hagisawa S, Nitta A, Hirabayashi H, Suzumura H, Arisaka O. Long-term survival of full trisomy 13 in a 14 year old male: a case report. Eur Rev Med Pharmacol Sci. 2016 Mar;20(5):919-22. PMID: 27010151.
- Springett A, Wellesley D, Greenlees R, Loane M, Addor MC, Arriola L, Bergman J, Cavero-Carbonell C, Csaky-Szunyogh M, Draper ES, Garne E, Gatt M, Haeusler M, Khoshnood B, Klungsoyr K, Lynch C, Dias CM, McDonnell R, Nelen V, O’Mahony M, Pierini A, Queisser-Luft A, Rankin J, Rissmann A, Rounding C, Stoianova S, Tuckerz D, Zymak-Zakutnia N, Morris JK. Congenital anomalies associated with trisomy 18 or trisomy 13: A registry-based study in 16 European countries, 2000-2011. Am J Med Genet A. 2015 Dec;167A(12):3062-9.
- Herold T, Metzeler KH, Vosberg S, Hartmann L, Röllig C, Stölzel F, Schneider S, Hubmann M, Zellmeier E, Ksienzyk B, Jurinovic V, Pasalic Z, Kakadia PM, Dufour A, Graf A, Krebs S, Blum H, Sauerland MC, Büchner T, Berdel WE, Woermann BJ, Bornhäuser M, Ehninger G, Mansmann U, Hiddemann W, Bohlander SK, Spiekermann K, Greif PA. Isolated trisomy 13 defines a homogeneous AML subgroup with high frequency of mutations in spliceosome genes and poor prognosis. Blood. 2014 Aug 21;124(8):1304-11. doi: 10.1182/blood-2013-12-540716. Epub 2014 Jun 12. PMID: 24923295.
- Plaiasu V, Ochiana D, Motei G, Anca I, Georgescu A. Clinical relevance of cytogenetics to pediatric practice. Postnatal findings of Patau syndrome – Review of 5 cases. Maedica (Bucur). 2010 Jul;5(3):178-85. PMID: 21977150; PMCID: PMC3177545.
- Dicker F, Haferlach C, Kern W, Haferlach T, Schnittger S. Trisomy 13 is strongly associated with AML1/RUNX1 mutations and increased FLT3 expression in acute myeloid leukemia. Blood. 2007 Aug 15;110(4):1308-16. doi: 10.1182/blood-2007-02-072595. Epub 2007 May 7. PMID: 17485549.
- Hall HE, Chan ER, Collins A, Judis L, Shirley S, Surti U, Hoffner L, Cockwell AE, Jacobs PA, Hassold TJ. The origin of trisomy 13. Am J Med Genet A. 2007 Oct 1;143A(19):2242-8. doi: 10.1002/ajmg.a.31913. PMID: 17853475.
- Pont SJ, Robbins JM, Bird TM, Gibson JB, Cleves MA, Tilford JM, Aitken ME. Congenital malformations among liveborn infants with trisomies 18 and 13. Am J Med Genet A. 2006 Aug 15;140(16):1749-56. doi: 10.1002/ajmg.a.31382. PMID: 16835915.
- Moerman P, Fryns JP, van der Steen K, Kleczkowska A, Lauweryns J. The pathology of trisomy 13 syndrome. A study of 12 cases. Hum Genet. 1988 Dec;80(4):349-56.
- Tharapel SA, Lewandowski RC, Tharapel AT, Wilroy RS Jr. Phenotype-karyotype correlation in patients trisomic for various segments of chromosome 13. J Med Genet. 1986 Aug;23(4):310-5. doi: 10.1136/jmg.23.4.310. PMID: 3746829; PMCID: PMC1049695.
- Hook EB. Rates of chromosome abnormalities at different maternal ages. Obstet Gynecol 1981 , 58, 282-5. PMID: 6455611
- Hook EB, Cross PK & Schreinemachers DM. Chromosomal abnormality rates at amniocentesis and in live-born infants. JAMA 1983 , 249, 2034-8. PMID: 6220164
- Schreinemachers DM, Cross PK & Hook EB. Rates of trisomies 21, 18, 13 and other chromosome abnormalities in about 20 000 prenatal studies compared with estimated rates in live births. Hum. Genet. 1982 , 61, 318-24. PMID: 6891368.
- Patau K, Smith DW, Therman E, Inhorn SL, Wagner HP. Multiple congenital anomaly caused by an extra autosome. Lancet. 1960; 1(7128): 790‐793.
- Rekomendacje Zespołu Ekspertów Polskiego Towarzystwa Ginekologicznego oraz Polskiego Towarzystwa Genetyki Człowieka w zakresie przesiewowego badania genetycznego wykonanego na wolnym płodowym DNA, Ginekologia i Perinatologia Praktyczna 2017, tom 2, nr 5, 230–233.
- Rekomendacje Sekcji Ultrasonografii Polskiego Towarzystwa Ginekologów i Położników w zakresie przesiewowej diagnostyki ultrasonograficznej w ciąży o przebiegu prawidłowym – 2020 r.