Disomia jednorodzicielska i imprinting genomowy

Spis treści
Co to jest disomia jednorodzicielska?
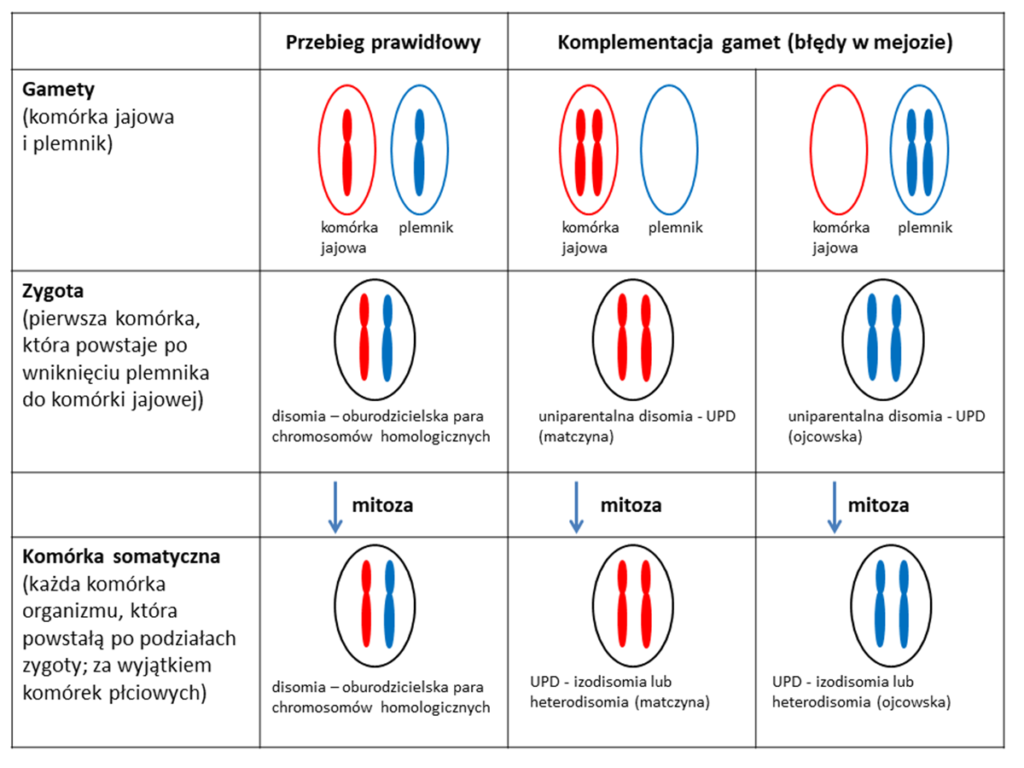
Disomia jednorodzicielska (UPD, ang. uniparental disomy) to obecność w komórkach pary chromosomów homologicznych (tej samej pary) pochodzących tylko od jednego z rodziców – matki lub ojca. Teoretyczny koncept disomii jednorodzicielskiej został stworzony w 1980 roku, a jego autorem jest szwajcarski lekarz i genetyk Eric Engel. Co ciekawe, dopiero 7 lat później teoria ta została potwierdzona w badaniach wykorzystujących techniki biologii molekularnej.
Wiemy, że człowiek jest organizmem diploidalnym, czyli w jego komórkach znajduje się podwójny zestaw chromosomów homologicznych. Po zapłodnieniu (komórka jajowa łączy się z plemnikiem) powstaje zygota, czyli pierwsza komórka, która po kolejnych podziałach komórkowych da nowy organizm. Właśnie dzięki rozmnażaniu płciowemu (do powstania potomstwa potrzebne jest połączenie się komórki rozrodczej żeńskiej i męskiej), człowiek posiada 23 pary chromosomów homologicznych, przy czym w każdej parze jeden chromosom pochodzi od matki, a drugi od ojca.
Wyróżniamy 2 rodzaje disomii jednorodzicielskiej:
- Heterodisomia uniparentalna/jednorodzicielska (UPHD, ang. uniparental heterodisomy) – w komórce występują 2 chromosomy homologiczne różniące się od siebie (2 różne kopie), ale oczywiście pochodzące od jednego z rodziców; heterodisomia jest wynikiem nondysjunkcji (nieprawidłowego rozejścia się) chromosomów homologicznych podczas pierwszego podziału mejotycznego (GAMETOGENEZA).
- Izodisomia uniparentalna/jednorodzicielska (UPID, ang. uniparental isodisomy) – w komórce występują 2 identyczne chromosomy homologiczne, czyli innymi słowy są to 2 identyczne kopie tego samego chromosomu; izodisomia jest wynikiem nondysjunkcji chromatyd siostrzanych podczas drugiego podziału mejotycznego.

Co to jest imprinting genomowy, czyli skąd wiadomo, że w danej parze chromosomów homologicznych, jeden jest pochodzenia matczynego, a drugi ojcowskiego?
Imprinting genomowy (inaczej piętnowanie genomowe lub piętnowanie rodzicielskie) to jeden z mechanizmów epigenetycznej modyfikacji ekspresji genów w komórce. Czynniki epigenetyczne to takie czynniki, które mogą zmieniać ekspresję genów (czyli modyfikować genom) bez zmian w sekwencji nukleotydowej DNA (materiału genetycznego). Jednym z takich czynników epigenetycznych jest metylacja kwasu deoksyrybonukleinowego (DNA). Proces ten polega na przyłączeniu do DNA grupy metylowej (a konkretnie do cytozyny, czyli jednej z zasad pirymidynowych budujących DNA), co zmienia aktywność genu, w sekwencji którego doszło do metylacji. Zmiany epigenetyczne mogą aktywować lub wyłączać określone geny, mogą również precyzować, które białka będą produkowane. Metylacja DNA jest m.in. odpowiedzialna za tzw. piętnowanie (imprinting) gamet (komórek rozrodczych). Oznacza to, że gameta żeńska (komórka jajowa) i gameta męska (plemnik) mają różniące się od siebie wzory metylacji. Zygota, czyli pierwsza komórka, która powstaje w wyniku zapłodnienia posiada więc materiał genetyczny obu gamet (a zatem obojga rodziców), który jest „znakowany” odmiennym wzorem metylacji. Zygota ulegając kolejnym podziałom komórkowym przekazuje ten wzór metylacji (równocześnie matczyny i ojcowski) nowym komórkom potomnym, które tworzą nowy organizm. Innymi słowy – każda komórka człowieka wywodzi się z zygoty i zawiera taki sam materiał genetyczny jak zygota. Tak więc w każdej komórce znajduje się zestaw 23 par chromosomów homologicznych, z których połowa pochodzi od matki i ma swój własny wzór metylacji, a druga połowa od ojca, która również ma swój własny wzór metylacji.
Dzięki temu analizując materiał genetyczny, możemy stwierdzić czy w danej parze chromosomów homologicznych w komórce jeden z chromosomów pochodzi od ojca, a drugi od matki. U człowieka imprinting genomowy, czyli to „nałożenie piętna” w postaci metylacji DNA zachodzi podczas gametogenezy, na etapie replikacji (kopiowania) DNA podczas podziału mejotycznego. Co ważne, aby powstające gamety mogły zostać „oznakowane”, najpierw musi zostać usunięty natywny (macierzysty) wzór metylacji. W tym momencie proces ten wydaje się zagmatwany i zrozumienie imprintingu genomowego może być trudne. Opierając się na przykładzie możemy wytłumaczyć to następująco: kobieta, przyszła mama we wszystkich swoich komórkach posiada zestaw 23 par chromosomów, które mają wzór metylacji po rodzicach tej kobiety. Ale komórki jajowe tej kobiety na etapie pierwotnych komórek macierzystych (oogoniów) muszą zostać oznaczone na nowo, już zgodnie z „płcią” komórek macierzystych gamet. Dlaczego? Ponieważ komórka jajowa ma haploidalny, pojedynczy zestaw chromosomów (23 chromosomy), który razem z haploidalnym zestawem chromosomów plemnika po zapłodnieniu stworzy pełen garnitur chromosomów (23 pary chromosomów) pierwszej komórki nowego organizmu, czyli zygoty. W związku z tym w takiej komórce jajowej powstaje nowy wzór metylacji, który znakuje ten haploidalny zestaw chromosomów jako chromosomy matczyne. Taka sama sytuacja dotyczy plemników. Aby to było możliwe, pierwszym etapem jest usunięcie, wymazanie rodzicielskiego wzoru metylacji (tzw. demetylacja) po rodzicach w oogoniach i spermatogoniach, czyli komórkach macierzystych gamet. Następnie w procesie gametogenezy, podczas rozwoju gamet na ich chromosomy zostaje nałożony nowy wzór metylacji. Czyli po skończonym podziale mejotycznym wszystkie plemniki posiadają imprinting (wzór metylacji) odojcowski, a komórki jajowe imprinting odmatczyny. Dzięki temu po połączeniu się gamety żeńskiej i męskiej – w zygocie, znajdziemy po jednym chromosomie odojcowskim i odmatczynym danej pary chromosomów homologicznych. Nie wszystkie geny są piętnowane przez całe nasze życie. Większość z nich ulega gwałtownej demetylacji zanim zarodek osiągnie stadium moruli (czyli gdy składa się z 12-16 komórek, co ma miejsce w ok. 4 dobie po zapłodnieniu). Genom ojcowski ulega demetylacji szybciej niż genom matczyny. Proces ten nie dotyczy pewnej grupy genów, które posiadają rodzicielski wzór metylacji (czyli tzw. piętno rodzicielskie) przez całe nasze życie. Ten wzór piętna (metylacji) odziedziczony od rodziców nie ulega zmianom w trakcie rozwoju organizmu.

Warto również nadmienić, że proces metylacji DNA zachodzi nie tylko w procesie gametogenezy. Zmiany epigenetyczne mogą zachodzić przez całe nasze życie (zarówno pre-, jak i postnatalne), a coraz więcej doniesień mówi również o tym, że mogą być one również utrwalane i dziedziczone przez kolejne pokolenia. Szczególną uwagę zwraca się na tzw. programowanie wewnątrzmaciczne, które wiąże wpływ różnych niekorzystnych czynników środowiskowych podczas ciąży na powstanie zmian epigenetycznych DNA u potomstwa, których konsekwencją mogą być wady strukturalne i rozwojowe oraz zaburzenia zdrowotne u potomstwa. Coraz mocniej podkreśla się ważną rolę czynników epigenetycznych w patomechanizmie tzw. chorób wieloczynnikowych (np. otyłość, choroby układu krążenia, choroby metaboliczne, choroby nowotworowe, choroby psychiczne czy też choroby neurodegeneracyjne). Wśród zidentyfikowanych czynników środowiskowych wpływających na zmiany epigenetyczne są m.in. żywienie, stres, używki (w tym tytoń i alkohol) czy też ekspozycja na niektóre substancje chemiczne. Takim bardzo podstawowym przykładem wpływu czynników środowiskowych na epigenetykę jest właściwa podaż folianów w trakcie ciąży (jest ona niezmiernie ważna dla prawidłowości przebiegu procesu metylacji w mechanizmie piętnowania/imprintingu genomowego). W tym miejscu chcieliśmy jedynie zasygnalizować temat zmian epigenetycznych i ich wpływu na nasze zdrowie, gdyż w pewnym stopniu wiąże się on z kwestiami związanymi z disomią jednorodzicielską. Z drugiej strony problematyka związana z epigenetyką jest na tyle ciekawa, a jednocześnie obszerna, że wymaga oddzielnego na nią spojrzenia i opracowania.
Jakie znaczenie ma obecność genów piętnowanych w genomie człowieka?
U ludzi dotychczas odnotowano istnienie ponad 80 genów piętnowanych. Geny te pełnią ważne funkcje rozwojowe, a zaburzenia piętnowania, czy też utrata tego piętna wiąże się z występowaniem szeregu chorób genetycznych, a także z niektórymi procesami nowotworzenia. Co ważne, rodzicielski imprinting genomowy nie jest rozłożony równomiernie na wszystkich chromosomach. Większość piętnowanych genów leży zgrupowana w zespoły (klastry) na chromosomach, 7, 11, 14, 15, 16 i 20. W obrębie tych zespołów znajduje się zwykle co najmniej jeden gen, który podlega ekspresji z allelu matczynego oraz jeden gen, podlegający ekspresji z allelu ojcowskiego (allele to warianty tego samego genu znajdujące się na chromosomach homologicznych). Dodatkowo ekspresja tych genów jest regulowana i koordynowana przez specjalne regiony DNA znajdujące się pomiędzy piętnowanymi genami, są to tzw. regiony ICR – rejony kontrolne piętnowania (ang. imprinting control region), nazywane również rejonami o zróżnicowanej metylacji albo centrami piętnowania. Rejony ICR są odpowiedzialne za ustalenie zróżnicowanego piętna i jego utrzymanie podczas rozwoju.
Rola imprintingu genomowego nie jest jeszcze do końca poznana. Przypuszcza się, że jedną z funkcji piętnowania genomowego może być zapobieganie homozygotyczności, np. partenogenezie (dzieworództwu). Wiadomo również, że różnice we wzorach metylacji genomu w zależności od płci są istotne dla odpowiedniego rozwoju organizmu począwszy od zapłodnienia. W badaniach przeprowadzonych na zarodkach mysich wykazano, że aby zarodek rozwijał się prawidłowo konieczna jest jednoczesna obecność genomów męskiego i żeńskiego, które różnią się w funkcjonowaniu. Po wniknięciu plemnika do komórki jajowej zanim materiał genetyczny z obu gamet połączy się, jądro plemnika przekształca się w tzw. przedjądrze męskie, a jądro komórki jajowej w przedjądrze żeńskie. W doświadczeniach sprawdzano jak rozwinie się zygota, która będzie posiadała dwa przedjądrza męskie lub dwa przedjądrza żeńskie, czyli będzie pozbawiona materiału genetycznego od jednego z rodziców. Okazało się, że w obu przypadkach rozwój takich zygot jest zaburzony. Zygoty androgenetyczne (2 przedjądrza męskie) miały nadmiernie rozwinięte błony płodowe i łożysko, a upośledzone struktury budowy zarodka. Rozwój zarodka był bardzo upośledzony i dochodziło do poronienia. Natomiast zygoty gynogenetyczne (2 przedjądrza żeńskie) charakteryzował niedorozwój łożyska, a za to intensywny rozwój zarodka. Taka sytuacja również kończyła się poronieniem, najprawdopodobniej z powodu niedożywienia zarodka. W badaniach tych wykazano więc, że genom ojcowski w dużym stopniu jest odpowiedzialny za wczesny rozwój łożyska, natomiast matczyny za rozwój zarodka, a przede wszystkim, że oba genomy są niezbędne dla rozwoju potomstwa.

W przypadku większości genów autosomalnych allele ojca i matki są równocenne. Oznacza to, że nie ma różnic w potencjale ekspresji między rodzicielskimi wariantami tego samego genu (matczynego i ojcowskiego). Nie jest tak w przypadku genów piętnowanych. Gen piętnowany jest nieaktywny, co powoduje, że ekspresji ulega tylko jeden allel rodzicielski – pochodzący od matki lub od ojca, a nie oba jednocześnie. Nabiera to znaczenia w przypadku kiedy aktywny allel ulegnie mutacji. Wtedy dochodzi to takiej sytuacji, w której dany gen na jednym chromosomie „nie działa” gdyż jest wyciszony poprzez imprinting genomowy, a na drugim z chromosomów homologicznych „nie działa” gdyż uległ mutacji, co może być przyczyną np. choroby genetycznej. Najbardziej znanymi chorobami genetycznymi, których przyczyna jest związana z imprintingiem genomowym są zespół Angelmana i zespół Pradera-Williego – w obu przypadkach napiętnowane geny znajdują się na chromosomie 15:
- Zespół Angelmana (ZESPÓŁ ANGELMANA) – genem napiętnowanym jest gen UBE3A, który leży na chromosomie 15 w regionie 15q11.2-q13. Gen ten ulega ekspresji tylko w mózgu i tylko z genu na chromosomie pochodzącym od matki, natomiast ojcowska kopia tego genu jest pozostaje wyciszona gdyż podlega imprintingowi genomowemu. Jeżeli dojdzie do delecji (utraty) materiału genetycznego regionu 15q11.2-q13 na chromosomie 15 pochodzącym od matki to taki organizm nie ma wówczas aktywnej kopii tego genu. Powoduje to chorobę neurogenetyczną zwaną zespołem Angelmana. Delecja regionu 15q11.2-q13 jest najczęstszą przyczyną tej wady genetycznej (65-75%).
- Zespół Pradera-Williego (ZESPÓL PRADERA – WILLIEGO) – napiętnowana jest grupa genów, która również leży na chromosomie 15 w regionie 15q11.2-q13 (m.in. gen MKRN3, MAGEL2, NDN, SNURF-SNRPN), ale w tym przypadku imprinting dotyczy chromosomu matczynego (na tym chromosomie geny te są wyciszone). Delecja ojcowskiego regionu 15q11.2-q13 spowoduje, że w danym organizmie nie będzie aktywnej wersji tych genów, co wywoła chorobę genetyczną o nazwie zespół Pradera-Williego. Delecja regionu 15q11.2-q13 jest najczęstszą przyczyną tej wady genetycznej i dotyczy ok. 70% przypadków.
Konsekwencje kliniczne imprintingu genomowego obserwujemy również w przypadku wystąpienia disomii jednorodzicielskiej chromosomów, o czym napiszemy poniżej.
Jak dochodzi do powstania disomii jednorodzicielskiej?
Mechanizm powstania disomii jednorodzicielskiej jest związany z naprawą aneuploidii, która powstała w wyniku błędów w procesie mejozy (podczas powstawania gamet) lub w procesie mitozy (już po zapłodnieniu). Innymi słowy, disomia jednorodzicielska jest konsekwencją powstania nieprawidłowych komórek rozrodczych lub nieprawidłowych komórek po podziale zygoty bądź blastomerów (komórek potomnych zygoty).
Mechanizmy powstania UPD, które są związane z nieprawidłową segregacją chromosomów w czasie mejozy
- Naprawa (korekta) trisomii (ang. trisomy rescue) – jeżeli w trakcie podziału mejotycznego powstanie nieprawidłowa gameta – diploidalna, zamiast haploidalnej, czyli taka, która posiada dwa chromosomy homologiczne (chromosomy tej samej pary) zamiast jednego chromosomu homologicznego, to po zapłodnieniu powstanie zygota trisomiczna (posiadająca trzy kopie chromosomów, zamiast dwóch). Zygota trisomiczna będzie więc posiadała dwa chromosomy homologiczne od jednego rodzica, a trzeci od drugiego rodzica. W takiej sytuacji mogą zostać uruchomione mechanizmy naprawcze, które doprowadzą do utraty jednej z tych trzech kopii chromosomów, aby uzyskać prawidłową komórkę diploidalną. Efektem takiej naprawy może być usunięcie tej kopii chromosomu, która pochodzi od jednego rodzica, a pozostawienie dwóch kopii od drugiego rodzica, co spowoduje powstanie disomii jednorodzicielskiej. Może to być zarówno heterodisomia, jak i izodisomia jednorodzicielska, w zależności od tego w którym podziale mejotycznym nastąpił błąd rozejścia chromosomów. Naprawa stanu trisomicznego, czyli postzygotyczne usunięcie dodatkowej (trzeciej) kopii chromosomu jest najczęstszą przyczyną powstania disomii jednorodzicielskiej (UPD). Badania wskazują, że ryzyko powstania disomii jednorodzicielskiej dla chromosomów 16 i 22 pary na drodze korekty trisomii wynosi 30%. W literaturze znajdziemy również opisy przypadków UPD chromosomów 7, 9, 10 i 15 pary.

- Naprawa (korekta) trisomii będącej konsekwencją translokacji Robertsonowskiej u rodzica – jeżeli jeden z rodziców jest nosicielem translokacji Robertsonowskiej (dwa chromosomy akrocentryczne pękają w miejscu centromeru i ich długie ramiona łączą się ze sobą), to w procesie mejozy może powstać gameta, która posiada dwa chromosomy homologiczne – jeden prawidłowy i drugi, który w wyniku translokacji został przyłączony do innego chromosomu). Taka gameta po zapłodnieniu da zygotę trisomiczną. Naprawa stanu trisomicznego (usunięcie dodatkowej kopii chromosomu) może być przyczyną heterodisomii jednorodzicielskiej. Innym możliwym przypadkiem jest powstanie translokacji Robertsonowskiej de novo podczas gametogenezy, co również prowadzi do powstania zygoty trisomicznej i w takim samym mechanizmie korekty trisomii może powstać UPD ( disomia jednorodzicielska).

- Naprawa (korekta) monosomii (ang. monosomy rescue) – analogicznie jak w przypadku naprawy trisomii, jeżeli w trakcie błędu i nieprawidłowego rozdziału chromosomów w podziale mejotycznym powstanie nieprawidłowa gameta, która nie posiada ani jednej kopii danego chromosomu (gameta nullisomiczna), to po zapłodnieniu prawidłową gametą powstanie zygota z monosomią tego chromosomu. Może wtedy dojść do uruchomienia systemów naprawczych, których celem jest wyrównanie takiego niezrównoważenia chromosomów, czyli do duplikacji monosomicznego chromosomu. Efektem takich działań będzie powstanie izodisomii jednorodzicielskiej. Taki mechanizm powstawania UPD nie jest częsty, w literaturze najbardziej znane są opisane przypadki izodisomii chromosomu 7 u dzieci z mukowiscydozą i niedoborem wzrostu.

- Komplementacja gamet – może zdarzyć się taka sytuacja, że w wyniku błędów w podziale mejotycznym u jednego rodzica powstanie tzw. gameta nullisomiczna (nieposiadająca chromosomu danej pary, a w skrajnych przypadkach pusta, czyli w ogóle bez chromosomów), a u drugiego rodzica w wyniku błędów w podziale mejotyczna powstanie gameta disomiczna (posiadająca dwie, zamiast jednej kopii danego chromosomu). Efektem połączenia (zapłodnienia) takich dwóch gamet: nullisomicznej i disomicznej będzie zygota z disomią jednorodzicielską. Oczywiście opisana sytuacja może mieć miejsce, jeżeli nullisomia i disomia dotyczy tej samej pary chromosomów w obu gametach. Komplementacja gamet zdarza się rzadko, dotychczas w literaturze opisano kilka takich przypadków. Takim skrajnym przykładem obrazującym komplementację gamet jest zaśniad groniasty, który powstaje w wyniku połączenia pustego oocytu (brak chromosomów) z dwoma plemnikami. Wtedy mamy do czynienia z disomią jednorodzicielską wszystkich chromosomów. Z takiej zygoty nie rozwinie się zarodek.
Postzygotyczne mechanizmy powstania UPD
- Nieprawidłowe rozejście się chromosomów podczas mitozy (mitoza to podziały komórkowe, którym ulega zygota, a następnie jej komórki potomne), które są związane z błędami w procesie mitozy – podobnie jak na etapie tworzenia się gamet, może dojść do nondysjunkcji chromosomów potomnych i późniejszej korekty błędu – usuwania lub duplikacji chromosomu, co prowadzi do izodisomii jednorodzicielskiej. Izodisomia jednorodzicielska, która powstała w ten sposób najczęściej występuje w mozaice z prawidłowymi komórkami, w których wszystkie chromosomy homologiczne mają oburodzicielskie pochodzenie, czyli w organizmie część komórek ma izodisomię, a część nie. Dzieje się tak dlatego, że do nondysjunkcji dochodzi już po zapłodnieniu, w trakcie kolejnych podziałów komórkowych. Innymi słowy – zygota ma wszystkie chromosomy homologiczne oburodzicielskie. Gdy zaczyna się dzielić na komórki potomne, na którymś etapie może dojść do błędu – nieprawidłowego rozejścia się chromosomów. Izodisomię jednorodzicielską będą miały tylko te komórki, które będą pochodziły od tej pierwszej komórki potomne, w której zaszła nondysjunkcja w trakcie mitozy.

- Crossing over w trakcie mitozy – prawidłowo crossing over, czyli wymiana materiału genetycznego pomiędzy chromatydami siostrzanymi w celu zachowania zmienności genetycznej ma miejsce w mejozie, czyli na etapie powstawania komórek jajowych i plemników. Może się zdarzyć (rzadko), że w trakcie mitozy dojdzie do pęknięcia nici DNA i „zgubienia” fragmentu chromosomu. W takiej sytuacji, w ramach naprawy nastąpi dobudowanie brakującego elementu materiału genetycznego poprzez skopiowanie go z drugiego homologicznego chromosomu. W ten sposób powstanie częściowa izodisomia jednorodzicielska, czyli w komórce będzie para chromosomów homologicznych pochodząca od obu rodziców, ale jeden z chromosomów będzie miał fragmenty takie same jak chromosom od drugiego rodzica.

Negatywne skutki disomii jednorodzicielskiej
Częstotliwość disomii jednorodzicielskiej wynosi 3 na 10000 urodzeń. Badania wykazują, że prawie trzykrotnie częściej występuje disomia matczyna niż ojcowska. Disomia jednorodzicielska może dotyczyć każdego z chromosomów, chociaż nie zawsze będzie się to wiązało z konsekwencjami klinicznymi UPD. Disomia jednorodzicielska chromosomów 6, 7, 11, 14, 15, 20 prowadzi do wrodzonych wad genetycznych.
Negatywne skutki disomii jednorodzicielskiej występują gdy:
- Chromosom, którego dotyczy disomia posiada geny piętnowane – takie przypadki chorób wynikających z UPD są najczęstsze. W takiej sytuacji w komórkach znajdują się 2 kopie genu, który jest piętnowany, a więc jest nieaktywny, a brakuje allelu, który podlega ekspresji, co powoduje określoną chorobę, niestety najczęściej z poważnymi objawami, do których należą m.in. wady wrodzone, wady dysmorficzne czy też niepełnosprawność intelektualna. W poniższej tabeli zostały zebrane najczęściej występujące choroby genetyczne wynikające z disomii jednorodzicielskiej chromosomów z imprintingiem genomowym.
UPD | Rodzic, od którego pochodzi piętno | Choroba genetyczna lub obraz kliniczny | Częstotliwość występowania | W jakim % przypadków UPD jest przyczyną choroby | Charakterystyczne objawy choroby |
UPD15 | matczyne | Zespół Pradera-Willego | 1/10000 -1/25000 | 20-25% | hipotonia, trudności w karmieniu, nadmierne łaknienie, otyłość, zaburzenia funkcji poznawczych, hypogonadyzm, niskorosłość, małe dłonie i stopy, subtelna dysmorfia twarzoczaszki, zez, skolioza |
UPD15 | ojcowskie | Zespół Angelmana | 1/12000 – 1/20000 | 3-7% | znaczne opóźnienie w rozwoju, zaburzenia chodu / równowagi, opóźnienie rozwoju mowy, zaburzenia neurologiczne, dysmorfia twarzy |
UPD11 | ojcowskie | Zespół Beckwitha-Wiedemanna | 1/15000 | 20% | makrosomia płodu, przepuklina pępowinowa / przepuklina pępkowa/rozdzielenie mięśni brzucha, hipoglikemia okołourodzeniowa, powiększony język (makroglosja), hemihipertrofia (nierówny wzrost jednej części ciała względem drugiej), organomegalia, charakterystyczny wyraz twarzy, który normalizuje się w dorosłości, zagięcia płatka ucha i zagłębienia za uchem, naczyniak płaski lub inne malformacje naczyniowe |
UPD11 | matczyne | Zespół Silvera-Rusella | 1/75000 – 1/100000 | 60% | opóźniony wzrost wewnątrzmaciczny, asymetria i dysmorfia ciała, zahamowany rozwój czaszki, charakterystyczne rysy twarzy (w tym nisko osadzone uszy), nadmierna potliwość, hipoglikemia |
UPD7 | matczyne | Zespół Silvera-Rusella | 1/75000 – 1/100000 | 10% | |
UPD14 | ojcowskie | Zespół Kagami–Ogata (KOS, zespół Wang) | Brak danych | 65% | wielowodzie, wady w obrębie brzucha i klatki piersiowej, podwyższona urodzeniowa masa ciała (hypertrofia mięśniowa), niepełnosprawność intelektualna w stopniu głębokim |
UPD14 | matczyne | Zespół Temple | Brak danych | 29% | przed- i pourodzeniowe zahamowanie wzrostu, wrodzona hipotonia, wiotkość stawów, opóźnienie rozwoju ruchowego, niepełnosprawność intelektualna w stopniu umiarkowanym |
UPD6 | ojcowskie | Przemijająca cukrzyca noworodków (TNDM) | 1/300000 | 41% | hiperglikemia, makroglosja, mogą współwystępować objawy neurologiczne, w tym padaczka, przeważnie ustępuje w wieku 18 miesięcy i nie wymaga dalszego leczenia, choć u ok. 50% pacjentów dochodzi do nawrotu choroby |
UPD20 | ojcowskie | Rzekoma niedoczynność przytarczyc 1b (zespół Albrighta, wrodzona osteodystrofia) | brak danych | 2-3% | hipokalcemia, hiperfosfatemia, zwiększone stężenie parathormonu (PTH we krwi oraz oporność narządów i tkanek docelowych na ten hormon |
UPD20 | matczyne | Zespół Mulchandani– | brak danych | 100% | przed- i pourodzeniowe zahamowanie wzrostu, nieprawidłowości budowy głowy, szyi, kończyn, moczowo—płciowego, nerwowego, mięśniowego, kostnego, powłok skórnych, wady układu pokarmowego skutkujące poważnymi problemami z odżywianiem |
- W wyniku disomii rodzic przekazuje dziecku 2 allele recesywne powodujące chorobę. Są to bardzo rzadkie przypadki wywołujące autosomalne choroby recesywne, które są przykładem dziedziczenia w sposób niemendlowski. Przypominamy, że sposób dziedziczenia cech autosomalnych (takich, których geny są zlokalizowane na autosomach, czyli chromosomach 1-22) może przebiegać w sposób dominujący lub recesywny. W przypadku chorób dziedziczonych w sposób dominujący, choroba ujawni się zarówno w układzie homozygotycznym, jak i heterozygotycznym, czyli wystarczy jeden allel danego genu, aby spowodować chorobę, za którą odpowiada. Natomiast w przypadku chorób dziedziczonych autosomalnie recesywnie choroba wystąpi tylko w układzie homozygotycznym, czyli potrzebne są 2 takie same allele. Pamiętając, że dziedziczymy po 1 allelu od każdego z obojga rodziców, oznacza to, że aby zachorować na chorobę autosomalną recesywną oboje z rodziców muszą być nosicielami takiego allelu. Wyjątkiem od tej zasady jest właśnie disomia jednorodzicielska, a właściwie izodisomia jednorodzicielska. Jeżeli dojdzie do sytuacji, że disomia dotyczy chromosomu, na którym znajduje się recesywny wariant genu odpowiedzialnego za daną chorobę lub gen uległ mutacji wywołującej chorobę, to w przypadku izodisomii , kiedy to mamy dwie identyczne kopie tego samego chromosomu mamy do czynienia z homozygotą. A więc występują dwa allele recesywne, co powoduje chorobę. Aktualnie w literaturze możemy znaleźć poniżej 100 opisów takich przypadków. Wśród dotychczas opisanych chorób autosomalnych recesywnych spowodowanych UPD, najczęściej odnotowywano te, które wynikają z disomii chromosomu 1, a najrzadziej te, które są związane z disomią chromosomu 9.
Diagnostyka
Diagnostyka chorób genetycznych będących wynikiem disomii jednorodzicielskiej (UPD) najczęściej odbywa się postnatalnie. Na podstawie objawów klinicznych zostaje postawione wstępne rozpoznanie, które ostatecznie weryfikuje się w badaniach genetycznych. W zależności od rodzaju rozpoznania genetyk dobiera odpowiednie badania, wśród nich mogą to być:
- fluorescencyjna hybrydyzacja in situ (FISH, ang. Fluorescent in situ Hybridization)
- porównawcza hybrydyzacja genomowa do mikromacierzy (aCGH, ang. array Comparative Genomic Hybridization)
- analiza metylacji w badanym regionie DNA (MS-MLPA , ang. Methylation-specific MLPA)
- badanie polimorfizmu krótkich powtórzeń tandemowych (STR, ang. Short Tandem Repeats)
- badanie polimorfizmu pojedynczego nukleotydu (SNP, ang. Single Nucleotide Polymorphism)
- badanie polimorfizmu długości fragmentów restrykcyjnych (RFLP, ang. restriction fragments length polymorphism)
- poszukiwanie mutacji w genach piętnowanych
W sytuacji, jeżeli wiadomo, że jedno z rodziców jest nosicielem translokacji Robertsonowskiej lub mutacji jednego z genów piętnowanych, można również przeprowadzić diagnostykę prenatalną. W tym celu wykonuje się inwazyjne badania prenatalne, jak amniopunkcja czy biopsja kosmówki. Dodatkowych informacji mogą również dostarczyć nieinwazyjne testy prenatalne (NIPT, ang. non-invasive prenatal testing), w których analizuje się wolne płodowe DNA (cffDNA, ang. cel free fetal DNA). Należy jednak pamiętać, że testy NIPT należą do prenatalnych badań przesiewowych, co oznacza, że na ich podstawie nie można postawić diagnozy. Wynik testu NIPT daje nam informację odnośnie wystąpienia ewentualnego ryzyka danej choroby genetycznej i może być przesłanką do dalszej diagnostyki w celu postawienia rozpoznania.
Bibliografia
- Scuffins J, Keller-Ramey J, Dyer L, Douglas G, Torene R, Gainullin V, Juusola J, Meck J, Retterer K. Uniparental disomy in a population of 32,067 clinical exome trios. Genet Med. 2021 Jun;23(6):1101-1107. doi: 10.1038/s41436-020-01092-8. Epub 2021 Jan 25. PMID: 33495530; PMCID: PMC8187148.
- Eggermann T. Prenatal Detection of Uniparental Disomies (UPD): Intended and Incidental Finding in the Era of Next Generation Genomics. Genes (Basel). 2020 Dec 3;11(12):1454. doi: 10.3390/genes11121454. PMID: 33287348; PMCID: PMC7761756.
- Del Gaudio D, Shinawi M, Astbury C, Tayeh MK, Deak KL, Raca G; ACMG Laboratory Quality Assurance Committee. Diagnostic testing for uniparental disomy: a points to consider statement from the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2020 Jul;22(7):1133-1141. doi: 10.1038/s41436-020-0782-9. Epub 2020 Apr 16. PMID: 32296163
- Nakka P, Pattillo Smith S, O’Donnell-Luria AH, McManus KF; 23andMe Research Team, Mountain JL, Ramachandran S, Sathirapongsasuti JF. Characterization of Prevalence and Health Consequences of Uniparental Disomy in Four Million Individuals from the General Population. Am J Hum Genet. 2019 Nov 7;105(5):921-932. doi: 10.1016/j.ajhg.2019.09.016. Epub 2019 Oct 10. PMID: 31607426; PMCID: PMC6848996.
- Xiao B, Wang L, Liu H, Fan Y, Xu Y, Sun Y, Qiu W. Uniparental isodisomy caused autosomal recessive diseases: NGS-based analysis allows the concurrent detection of homogenous variants and copy-neutral loss of heterozygosity. Mol Genet Genomic Med. 2019 Oct;7(10):e00945. doi: 10.1002/mgg3.945. Epub 2019 Aug 27. PMID: 31454184; PMCID: PMC6785455.
- Millership, S.J., Van de Pette, M. & Withers, D.J. Genomic imprinting and its effects on postnatal growth and adult metabolism. Cell. Mol. Life Sci. 76, 4009–4021 (2019). https://doi.org/10.1007/s00018-019-03197-z
- Niida Y, Ozaki M, Shimizu M, Ueno K, Tanaka T. Classification of Uniparental Isodisomy Patterns That Cause Autosomal Recessive Disorders: Proposed Mechanisms of Different Proportions and Parental Origin in Each Pattern. Cytogenet Genome Res. 2018;154(3):137-146. doi: 10.1159/000488572. Epub 2018 Apr 14. PMID: 29656286.
- Matsubara K, Kagami M, Fukami M. Uniparental disomy as a cause of pediatric endocrine disorders. Clin Pediatr Endocrinol. 2018;27(3):113-121. doi: 10.1297/cpe.27.113. Epub 2018 Jul 31. PMID: 30083028; PMCID: PMC6073059.
- Makishima H, Maciejewski JP. Pathogenesis and consequences of uniparental disomy in cancer. Clin Cancer Res. 2011 Jun 15;17(12):3913-23. doi: 10.1158/1078-0432.CCR-10-2900. Epub 2011 Apr 25. PMID: 21518781; PMCID: PMC3523887.
- Piedrahita JA. The role of imprinted genes in fetal growth abnormalities. Birth Defects Res A Clin Mol Teratol. 2011 Aug;91(8):682-92. doi: 10.1002/bdra.20795. Epub 2011 Jun 6. PMID: 21648055; PMCID: PMC3189628.
- Amor DJ, Halliday J. A review of known imprinting syndromes and their association with assisted reproduction technologies. Hum Reprod. 2008 Dec;23(12):2826-34. doi: 10.1093/humrep/den310. Epub 2008 Aug 14. PMID: 18703582.
- Kotzot D. Complex and segmental uniparental disomy (UPD): review and lessons from rare chromosomal complements. J Med Genet. 2001 Aug;38(8):497-507. doi: 10.1136/jmg.38.8.497. PMID: 11483637; PMCID: PMC1734925.
- Engel E, DeLozier-Blanchet CD. Uniparental disomy, isodisomy, and imprinting: probable effects in man and strategies for their detection. Am J Med Genet. 1991 Sep 15;40(4):432-9. doi: 10.1002/ajmg.1320400411. PMID: 1746607.
- Spence JE, Perciaccante RG, Greig GM, Willard HF, Ledbetter DH, Hejtmancik JF, Pollack MS, O’Brien WE, Beaudet AL. Uniparental disomy as a mechanism for human genetic disease. Am J Hum Genet. 1988 Feb;42(2):217-26. PMID: 2893543; PMCID: PMC1715272.
- Creau-Goldberg, N., Gegonne, A., Delabar, J., et al. Maternal origin of a de novo balanced t(21q21q) identified by ets-2 polymorphism. Hum Genet 1987; 76:396–398.