Co to jest delecja i zespół delecyjny
Delecja to rodzaj strukturalnej aberracji (nieprawidłowości) chromosomowej. Innymi słowy jest to rodzaj mutacji chromosomowej, gdzie zaburzona jest struktura chromosomu. Delecja to ubytek fragmentu chromosomu wraz z genami, które się w tym fragmencie znajdują. W wyniku utraty części materiału genetycznego powstają rzadkie, wrodzone choroby genetyczne nazywane ogólnie zespołami delecyjnymi. W zależności od tego, która część DNA zostaje utracona, wykształcają się charakterystyczne objawy, specyficzne dla danego zespołu delecyjnego.

Spis treści
Co oznacza delecja fragmentu chromosomu?
Wiemy, że prawidłowo, każda komórka ludzkiego ciała posiada zestaw 46 chromosomów układających się w 23 pary, a chromosomy w parze pochodzą jeden od matki, a drugi od ojca. Wyjątkiem są komórki rozrodcze, czyli komórka jajowa i plemniki, które posiadają pojedynczy zestaw, czyli 23 chromosomy. Po zapłodnieniu (połączeniu się komórki jajowej z plemnikiem) powstaje pierwsza komórka nowego organizmu, czyli zygota posiadająca już pełen zestaw chromosomów, a więc 23 pary. W kolejno następujących po sobie mitotycznych podziałach komórkowych (GAMETOGENEZA), z zygoty, a następnie komórek z niej powstałych, tworzą się kolejne nowe komórki budujące organizm potomny.
Każdy chromosom zbudowany jest z dwuniciowej cząsteczki DNA (kwasu deoksyrybonukleinowego) oraz białek. Informacja genetyczna zakodowana jest właśnie w chromosomach, w sekwencji nukleotydów DNA i przechowywana jest w jądrze każdej komórki jako tzw. genom jądrowy. W pojedynczym chromosomie może znajdować się od 50 do 250 milionów nukleotydów (pojedynczych cegiełek budujących DNA). Zestaw informacji dla organizmu, który warunkuje jego rozwój, występowanie poszczególnych cech zorganizowany jest w postaci genów. Gen (GEN) jest umowną jednostką dziedziczenia, w bardzo dużym uproszczeniu możemy powiedzieć, że jest to określona sekwencja DNA, która koduje daną cechę, proces, a najczęściej jego składową (niewiele cech dziedziczymy jednogenowo). W prawidłowo zbudowanym chromosomie, każdy gen posiada swoje stałe miejsce tzw. locus. Jedną z nieprawidłowości, która może dotknąć struktury (budowy) chromosomu jest delecja, czyli utrata fragmentu chromosomu. Jeżeli utrata obejmuje końcówki chromosomu, nazywana jest delecją terminalną, a gdy dochodzi do utraty środkowej części chromosomu, mówimy o delecji interstycjalnej.

Do delecji, a więc utraty części materiału genetycznego dochodzi w wyniku pęknięcia chromosomu. Do takiej mutacji strukturalnej może dojść na każdym z chromosomów. Odcinki niektórych chromosomów wykazują szczególną łamliwość. Należą do nich m.in. odcinek 13q chromosomu 2 (2q13), odcinek 27q chromosomu X (Xq13) oraz wiele miejsc na chromosomach 6, 9,12 i 20. Miejsca te są nazywane kruchymi miejscami genomu. Są one szczególnie wrażliwe na działanie mutagenów.
Do określenia lokalizacji ubytku DNA używa się skrótu „del” od słowa delecja oraz podaje się numer chromosomu, którego ona dotyczy, następnie wskazuje się na którym ramieniu jest delecja (p – ramię krótkie lub q – ramię długie), jaki jest numeru regionu i numer prążka, którego brakuje. Regiony leżące najbliżej centromeru (miejsca gdzie długie ramię łączy się z krótkim ramieniem) noszą numer 1, oddalające się w kierunku obwodowym – kolejne numery.
Dla przykładu przy delecji 17p13.2 utracony zostaje fragment 1 chromosomu z ramienia krótkiego, region 1, prążek 3, subprążek 2.
Delecja a mikrodelecja
Delecje mogą mieć również różną wielkość. W zależności od rozmiaru utraconego odcinka chromosomu, mówimy o delecji lub mikrodelecji. Mikrodelecja dotyczy małych zmian (mówimy o nich często małe delecje) – utrata sekwencji DNA o wielkości poniżej 5 Mpz (milionów par zasad). Utraty tak niewielkiego fragmentu chromosomu nie możemy zidentyfikować klasycznymi metodami cytogenetycznymi badania kariotypu (zbyt niska rozdzielczość badania). Do identyfikacji mikrodelecji wykorzystywane są molekularne metody cytogenetyki, o czym przeczytasz więcej poniżej w części dotyczącej diagnostyki chorób genetycznych.
Delecje a choroby genetyczne
W przypadku delecji, rozwijający się organizm nie posiada części chromosomu, a co za tym idzie, najczęściej nie posiada również części genów. Tak więc skutki fenotypowe, czyli objawy delecji wynikają z utraty informacji genetycznej. W zależności od tego którego chromosomu dotyczy delecja, czy i jakie geny zostały utracone, osoba dotknięta taką aberracją chromosomową może mieć różne problemy zdrowotne. Delecje powodują wiele różnych zaburzeń rozwojowych, a o stopniu nasilenia objawów decyduje miejsce i rozmiar obszaru DNA, który został utracony. Choroby genetyczne, które są wynikiem delecji zyskały wspólną nazwę zespołów delecyjnych, a w sytuacji jeżeli utracony odcinek chromosomu jest mniejszy niż 5 Mpz, wymiennie stosuje się określenie zespół mikrodelecyjny. Warto również wiedzieć o tym, że nie każda delecja czy mikrodelecja będzie miała niekorzystny wpływ na zdrowie. By doszło do wykształcenia choroby musi zostać utracony tzw. region krytyczny który definiuje się jako najmniejszy region danego chromosomu, którego utrata powoduje określony efekt kliniczny. Część strukturalnych zmian chromosomowych tego typu określana jest jako łagodna i nie wiąże się z występowaniem objawów chorobowych. Zdarzają się również delecje dużych odcinków chromosomowych, które z kolei zazwyczaj są letalne jeszcze na etapie wczesnej ciąży.
Wielkość delecji, a więc rozmiar utraconego odcinka chromosomu może różnić się w obrębie danego zespołu delecyjnego. Oznacza to, że zespół delecyjny nie jest jednorodny – różni się rozmiarem delecji, a co za tym idzie może również różnić się zestawem i nasileniem objawów choroby.
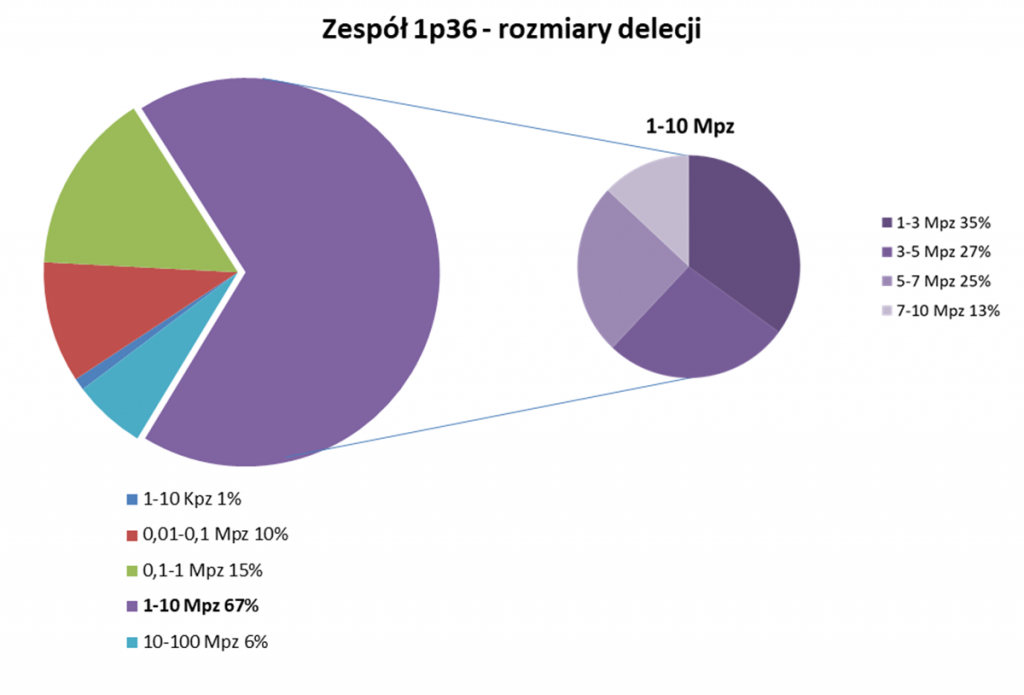
Możemy to przedstawić na przykładzie zespołu delecji / mikrodelecji 1p36, czyli choroby genetycznej, której przyczyną jest utrata części odcinka 36 chromosomu 1, który znajduje się na jego krótkim ramieniu. W przypadku tej delecji najmniejszy znany rozmiar utraconego fragmentu DNA wynosi 1,32 Kpz (tysiące par zasad), a największy znany rozmiar utraconego fragmentu DNA to 12,77 Mpz ( miliona par zasad). Widzimy więc, że w obrębie tej jednej wady genetycznej różnice są ogromne – od 1,32 Kpz do 12,77 Mpz, a więc ten zespół wad genetycznych może być zarówno delecją, jak i mikrodelecją. Ma to swoje implikacje zarówno w samej diagnostyce (np. przy wyborze technik o rozdzielczości 5-10 Mpz, możemy wykryć jedynie część przypadków tej delecji), jak i obrazie klinicznym choroby (różne symptomy i różne nasilenie choroby w zależności od tego jak duży odcinek i jakie geny zostały utracone).
Najczęstsze zespoły delecyjne i mikrodelecyjne
Najczęstsze zespoły delecyjne i mikrodelecyjne | Częstość występowania |
Zespół Di George’a (delecja22q11.2) | 1/4000 |
Zespół delecji 1p36 | 1/4000 – 1/10000 |
Zespół delecji proksymalnej 16p11.2 | 1/5000 |
Zespół Angelmana (delecja 15q11.2) | 1/12000 |
Zespół Pradera-Williego (delecja 15q11.2) | 1/10000 – 1/25000 |
Zespół Williams i Beurena (delecja 7q11.23) | 1/7500 – 1/20000 |
Zespół Smith-Magenis (delecja 17p11.2) | 1/15000 – 1/25000 |
Zespół Wolfa-Hirschorna (delecja 4p16.3) | 1/50000 |
Zespół Van der Woude (delecja 1q32-q41) | 1/40000 – 1/100000 |
Zespół Cri du Chat | 1/20000 – 1/50000 |
Jak dochodzi do utraty części DNA?
Na to pytanie nie ma jednoznacznej odpowiedzi. Do utraty fragmentu chromosomu, czyli do delecji może dojść na kilku etapach, m.in. podczas gametogenezy (powstawania komórek rozrodczych – odpowiednio komórki jajowej i plemników), ale także później w trakcie procesu łączenia komórek rozrodczych, czy podczas pierwszych podziałów komórkowych po zapłodnieniu. Często takie zmiany strukturalne chromosomów powstają niezależnie, de novo, nie można się przed nimi zabezpieczyć, a ich powstanie nie jest niczyją winą. Większość dzieci obarczonych tymi schorzeniami genetycznymi ma zupełnie zdrowych rodziców.
W przypadku niektórych chorób genetycznych, jak np. aneuploidie, czyli zaburzenia liczby chromosomów (np. trisomie, monosomie) czynnikiem zwiększającym ryzyko ich wystąpienia jest wiek kobiety w ciąży. W przypadku zmian submikroskopowych jakimi są mikrodelecje ryzyko ich wystąpienia nie zależy od wieku matki, co powoduje, że u dzieci kobiet poniżej 35 roku życia mogą one zdarzać się częściej niż np. zespół Downa (trisomia 21). Oczywiście warto przypomnieć (pisaliśmy o tym wyżej), że nie każda submikroskopowa utrata materiału genetycznego ma swoje odzwierciedlenie w postaci problemów zdrowotnych. Wszystko zależy od tego jaki region chromosomu został nią dotknięty.

Czy chorobę genetyczną wynikającą z delecji można odziedziczyć po rodzicach?
Istnieje taka możliwość, zwłaszcza w sytuacji jeżeli jeden z rodziców jest również obarczony taką wadą chromosomową. Ryzyko odziedziczenia danej wady genetycznej przez potomstwo zawsze rozpatrujemy indywidualnie i konsultujemy z lekarzem specjalistą.
W grupie ryzyka wystąpienia wad genetycznych, m.in. delecji u potomstwa są osoby z translokacją zrównoważoną lub inwersją. Inwersja to aberracja strukturalna, która polega na odwróceniu fragmentu chromosomu między dwoma miejscami złamań w chromosomie. Obecność inwersji zakłóca łączenie się w pary chromosomów homologicznych podczas mejozy, a więc może wpływać na powstawanie gamet z nieprawidłowym zestawem chromosomów zawierających duplikacje i delecje. Translokacja to rodzaj zmiany strukturalnej, która polega na tym, że fragment chromosomu ulega przemieszczeniu w inne miejsce tego samego lub innego chromosomu. A o translokacji zrównoważonej mówimy wtedy, gdy pomimo, że jakiś odcinek chromosomu znajduje się w nietypowym dla siebie miejscu, to materiał genetyczny nie został ani utracony ani dodany. Osoba z translokacją zrównoważoną ma nadal taką samą ilość DNA i najczęściej nie wpływa to na ekspresję genów oraz nie wiąże się to z wystąpieniem żadnych objawów chorobowych. O takich osobach mówimy, że są nosicielami translokacji. Są zdrowe, natomiast ich potomstwo ma większą szansę na otrzymanie chromosomu z brakującym fragmentem, a więc z niepełnym materiałem genetycznym, co może skutkować chorobą genetyczną. Jest to związane z tym jak przebiega proces gametogenezy (GAMETOGENEZA). Podczas powstawania komórek rozrodczych w procesie mejozy dochodzi do rozdzielenia homologicznych par chromosomów tak, aby komórka jajowa czy też plemnik miały nie 46, a 23 chromosomy. Podczas takiego rozdziału do komórki rozrodczej może trafić chromosom z brakującym odcinkiem, a równocześnie inny chromosom, do którego ten odcinek został przyłączony, nie trafi do tej samej gamety. Taki losowy rozdział chromosomów spowoduje, że powstanie komórka rozrodcza z brakującym DNA. Szacuje się, że 1 osoba na 500 posiada translokację zrównoważoną, w większości nie wiedząc o tym. Istnieje możliwość zbadania materiału genetycznego na obecność translokacji. Badanie polega na pobraniu niewielkiej ilości krwi i poddaniu zawartych w niej limfocytów analizie cytogenetycznej. Warto również pamiętać o tym, że jeżeli chodzi o zespoły delecyjne, to wiele z nich powstaje de novo, czyli nie są dziedziczone po rodzicach, a do mutacji dochodzi najczęściej na etapie gametogenezy lub w trakcie pierwszych podziałów komórkowych po zapłodnieniu.
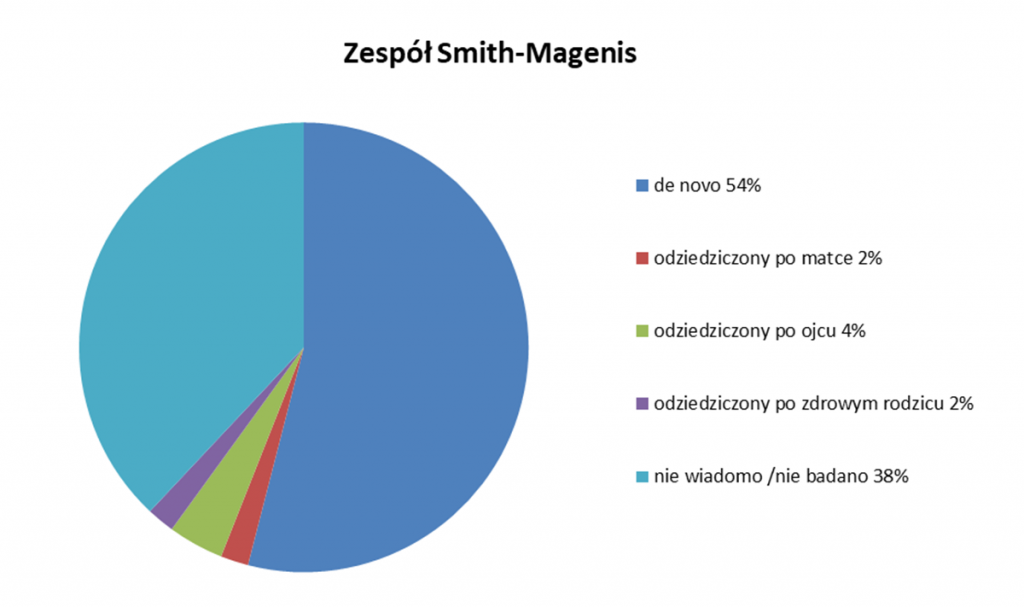
Dla każdej delecji udział poszczególnych czynników związanych z etiologią danej choroby genetycznej różni się. Możemy to prześledzić na poniższych grafikach, na przykładzie takich zespołów delecyjnych jak zespół Angelmana / Pradera-Williego i zespół Smith-Magenis. Widzimy, że np. w przypadku zespołu Smith-Magenis ponad połowa (54%) chorujących zyskała tą delecję de novo, czyli nie odziedziczyła jej po żadnym z rodziców. Z kolei w przypadku zespołu Angelmana / Pradera-Willliego delecja materiału genetycznego niezwiązana z dziedziczeniem, a powstała de novo to jedynie 15% chorujących.

Diagnostyka chorób genetycznych – zespoły delecyjne
W diagnostyce zespołów delecyjnych pomocne są badania genetyczne, które wykonywane są zarówno w okresie prenatalnym, jak i po urodzeniu dziecka (postnatalnie). Wszystko zależy od konkretnej sytuacji. Losowy charakter występowania delecji i mikrodelecji oraz ich z jednej strony różnorodność, a z drugiej rzadkość występowania utrudniają diagnostykę. W zależności od wielkości zespołu delecyjnego trzeba również dobrać badanie diagnostyczne o odpowiedniej rozdzielczości / czułości. Stąd też często diagnostyka w kierunku zespołów delecyjnych ma miejsce już po urodzeniu, kiedy odpowiedni zestaw badań diagnostycznych dobierany jest na podstawie objawów choroby u dziecka. W okresie ciąży, diagnostyczne badania prenatalne w kierunku delecji wykonywane są np. w sytuacji kiedy jeden z rodziców bądź ktoś z osób spokrewnionych jest chory lub jest nosicielem translokacji zrównoważonej, można wykonać badania w kierunku konkretnego schorzenia genetycznego.
Jak dzielimy badania prenatalne pozwalające wykryć takie zaburzenia genetyczne jak delecje u płodu:
- Nieinwazyjne badania prenatalne – należą do nich przede wszystkim badanie USG (USG PRENATALNE PIERWSZEGO TRYMESTRU) oraz test podwójny, czyli test PAPP-A (TEST PAPP-A), test potrójny (TEST POTRÓJNY W CIĄŻY) oraz testy NIPT badające cfDNA (CO TO JEST NIPT), np. test Harmony, test Nace, test Neobona, test NIFTY, test Nifty Pro, test Sanco, test Sanco Plus, test Veragene, test Veracity. Wymienione rodzaje nieinwazyjnych badań prenatalnych należą do testów przesiewowych, czyli takich, dzięki którym możemy ocenić ryzyko wystąpienia chorób genetycznych u płodu. Badania te są pierwszą linią pomocy w diagnostyce wad wrodzonych. Chociaż same w sobie nie są badaniami diagnostycznymi, a przesiewowymi, to ich czułość w zakresie wykrywania najczęstszych nieprawidłowości chromosomowych jest wysoka, a dodatkowo są to badania bezpieczne dla kobiety w ciąży i płodu. Wyniki pozytywne w tych testach powinny zostać zweryfikowane poprzez wykonanie badań diagnostycznych. Jeżeli chodzi o same zespoły delecyjne, to testy NIPT badające cfDNA są tymi, które są nakierowane na oznaczanie wybranych zespołów delecyjnych. Testy NIPT różnią się między sobą zakresem, a co za tym idzie również rozdzielczością badanych delecji. Niektóre z testów badają cały genom (wszystkie chromosomy) i oznaczają ryzyko wystąpienia u płodu dużych zespołów delecyjnych (zmiana równa lub wyższa 7 Mpz) jak np. test Nace, test Sanco czy też zespołów delecyjnych o wielkości 5 Mpz lub więcej, jak np. test Nifty, test Nifty Po. W przeciwieństwie do nich, testy NIPT jak test Harmony, test Panorama, test Sanco Plus, test Veragene, test Veracity nakierowane są na oznaczanie wybranych mikrodelecji, czyli submikroskopowych zmian w sekwencji DNA, gdzie utracony odcinek chromosomu jest mniejszy od 5 Mpz.
- Inwazyjne badania prenatalne – należą do nich amniopunkcja (AMNIOPUNKCJA)(do badania pobiera się płyn owodniowy), kordocenteza (KORDOCENTEZA)(do badania pobiera się krew z żyły pępowinowej), biopsja trofoblastu (BIOPSJA KOSMÓWKI)(pobranie fragmentu jednej z błon płodowych – kosmówki). Najczęściej wykonywanym inwazyjnym badaniem prenatalnym jest amniopunkcja. Z materiału pobranego w trakcie inwazyjnego badania prenatalnego izoluje się materiał genetyczny, który jest poddawany badaniom genetycznym. Rodzaje możliwych do wykonania testów cytogenetycznych znajdują się poniżej w części dotyczącej badań diagnostycznych, które można wykonać już po urodzeniu dziecka.
Diagnostyka zespołów delecyjnych i mikrodelecyjnych często ma miejsce już po porodzie. W zależności od objawów, które występują lekarz dobiera odpowiednie badanie cytogenetyczne. Jest to bardzo ważny etap, gdyż tak jak już wcześniej pisaliśmy – zmiany strukturalne chromosomów typu delecje, mikrodelecje są bardzo różnorodne, również pod względem rozmiaru utraconych odcinków DNA. To z kolei determinuje rodzaj badania diagnostycznego (ważna jest rozdzielczość takiego badania, czyli innymi słowy to jakiej wielkości zmiany w DNA można oznaczyć przy pomocy danej techniki cytogenetycznej).
Do podstawowych testów cytogenetycznych należą:
- Klasyczne badanie kariotypu – ocenia liczbę i strukturę wszystkich chromosomów. u. Stosowane obecnie techniki rutynowej analizy cytogenetycznej mają rozdzielczość około 5-10 Mpz (milionów par zasad).
- Fluorescencyjna hybrydyzacja in situ (FISH) – umożliwia wykrywanie delecji, duplikacji i rearanżacji niewielkich regionów w chromosomach. Analiza metodą FISH na obecność mikroduplikacji i mikrodelecji wymaga szczegółowych wskazań klinicznych, a więc znajomości regionu, w którym podejrzewa się występowanie zmian, gdyż to umożliwia wykorzystanie odpowiedniej sondy do detekcji zmiany strukturalnej. Jest wykorzystywana np. w sytuacji podejrzenia zespołu Di George’a czy zespołu Williamsa.
- Porównawcza hybrydyzacja genomowa do mikromacierzy, aCGH (arrayCGH, ang. array comparative genomic hybridization) – tą technikę cechuje lepsza rozdzielczość od tej, jaką można uzyskać za pomocą FISH oraz możliwość jednoczesnej oceny dużej liczby regionów chromosomowych pod kątem obecności delecji i duplikacji, czułość aCGH zależy od rodzaju zastosowanych sond, ale wykrywa zmiany genomu nawet rzędu 50-100 pz.
Zespół delecyjny – najczęściej zadawane pytania
Czy delecja i zespół delecyjny to to samo?
Delecja i zespół delecyjny to nie to samo, chociaż bywa, że oba terminy są stosowane wymiennie. Delecja to utrata fragmentu chromosomu, a zespół delecyjny to zespół objawów i wad wrodzonych spowodowany brakiem tego fragmentu chromosomu. Innymi słowy zespół delecyjny to choroba genetyczna spowodowana delecją fragmentu DNA.
Możemy to zobrazować przykładami:
- zespół delecji 1p36 to choroba genetyczna spowodowana utratą, czyli delecją fragmentu krótkiego ramienia chromosomu 1 (delecja może mieć różną wielkość i dotyczy regionu 3)
- zespół cri du chat (zespół kociego krzyku, kociego płaczu) to choroba genetyczna spowodowana utratą fragmentu krótkiego ramienia chromosomu 5 (wielkość delecji może być różna od fragmentu regionu 1 do całego ramienia krótkiego chromosomu 5)
Warto również dodać, że nie każda delecja ma charakter patologiczny. Niektóre delecje są neutralne dla zdrowia.
Co to jest zespół delecyjny?
Zespół delecyjny to wrodzona choroba genetyczna, która jest spowodowana delecją, czyli utratą fragmentu materiału genetycznego. Przykładem zespołu delecyjnego jest zespół DIGeorge’a, czyli najczęściej występujący zespół delecyjny. Jest on spowodowany delecją fragmentu długiego ramienia chromosomu 22 (delecja może mieć różną wielkość i dotyczy regionu 1)
Co to jest delecja?
Delecja jest to zmiana w materiale genetycznym, czyli w DNA, która polega na wypadnięciu (utracie) jego fragmentu. Wiemy, że podstawową cegiełką budującą kwasy nukleinowe, w tym DNA jest nukleotyd. Delecja może dotyczyć utraty jednego nukleotydu, kilku nukleotydów albo nawet kilku tysięcy czy milionów nukleotydów, co odpowiada większym fragmentom sekwencji DNA. Może więc obejmować fragment genu, jeden lub kilka genów.
Ile chromosomów posiada ludzka komórka?
Ludzkie komórki posiadają po 23 pary chromosomów (czyli po 46 chromosomów). Wyjątek stanowią gamety żeńskie i męskie, czyli komórki jajowe i plemniki, które mają po 23 chromosomy.
Czy każda komórka ludzkiego ciała ma 46 chromosomów?
Każda prawidłowa komórka ludzkiego ciała za wyjątkiem komórek rozrodczych ma 46 chromosomów. Komórki rozrodcze (gamety męskie i żeńskie, czyli komórki jajowe i plemniki) mają po 23 chromosomy.
Ile chromosomów ma plemnik?
Plemnik, czyli męska komórka rozrodcza ma 23 chromosomy. Wszystkie komórki rozrodcze człowieka, zarówno męskie, jak i żeńskie mają po 23 chromosomy, co stanowi połowę ogólnej ilości chromosomów, które mają pozostałe komórki ciała. Po wniknięciu plemnika do komórki jajowej powstaje zygota – nowa komórka, która ma 46 chromosomów. Jeżeli chcesz dowiedzieć się więcej o tym jak powstają plemniki, przeczytaj artykuł GAMETOGENEZA.
Ile chromosomów ma komórka jajowa?
Komórka jajowa (żeńska komórka rozrodcza) ma 23 chromosomy. Po połączeniu się komórki jajowej z plemnikiem powstaje zygota, czyli pierwsza komórka nowego organizmu, która ma 46 chromosomów. Z niej wyniku podziałów komórkowych powstaną kolejne komórki ludzkiego budujące ciało człowieka. Każda będzie miała po 46 chromosomów. Wyjątek stanowią plemniki i komórki jajowe, które mają połowę garnituru chromosomów, czyli 23. Jeżeli chcesz dowiedzieć się więcej jak dochodzi do tego, że komórki rozrodcze mają po 23 chromosomy, przeczytaj artykuł Gametogeneza.
Co to jest Locus?
Locus to określony obszar chromosomu, który zajmuje jeden gen. Tyle ile jest genów na danym chromosomie, tyle samo jest tam różnych loci.
Co to delecja terminalna?
Delecja terminalna to utrata końcowego odcinka ramienia chromosomu. Przykładem zespołu delecyjnego jest delecja terminalna długiego ramienia chromosomu 7. Jest to bardzo rzadka choroba genetyczna, która wiąże się z poważnym opóźnieniem rozwoju wewnątrzmacicznego płodu oraz rozwoju poporodowego. Wady związane są m.in. z niedorozwojem struktur mózgowia, dysmorfii twarzoczaszki, opóźnieniem wzrostu, uogólnioną hipotonią, niedosłuchem, zaburzeniami pracy serca, nieprawidłowym ukształtowaniem klatki piersiowej oraz wadami narządów płciowych u chłopców.
Co to delecja interstycjalna?
Delecja interstycjalna to utrata środkowego fragmentu chromosomu, czyli taka, która powstaje w wyniku dwóch pęknięć chromosomu, utraty środkowego fragmentu, a następnie powrotnego połączenia się pozostałych odcinków chromosomu. Przykładem choroby w wyniku delecji interstycjalnej jest zespół Pradera-Williego (delecja interstycjalna długiego ramienia chromosomu 15 pochodzenia ojcowskiego) oraz zespół Angelmana – delecja interstycjalna długiego ramienia chromosomu 15 pochodzenia matczynego (delecja dotyczy regionu 1).
Co to jest mikrodelecja?
Mikrodelecja to mała delecja, czyli utrata fragmentu DNA o wielkości do 5 Mpz (milionów par zasad). Przykładem choroby wywołanej mikrodelecją jest zespół Di George’a. W 96% przypadków jest on spowodowany delecją o wielkości 1,5-3 Mpz (dochodzi do utraty 24-30 genów).
Czy delecja i mikrodelecja to to samo?
Delecja i mikrodelecja to utrata fragmentu materiału genetycznego. W zależności od wielkości tego fragmentu mówimy o mikrodelecji (jeżeli utracony fragment ma do 5 Mpz, czyli 5 milionów par zasad) i o delecji (jeżeli utracony fragment ma powyżej 5 Mpz). W ten sposób wygląda podział naukowy.
Często jednak w codziennym języku możemy spotkać się z ogólnym określeniem delecji bez względu na wielkość utraconego obszaru na chromosomie.
Czy ryzyko zespołu delecyjnego rośnie z wiekiem matki?
Nie, ryzyko zespołu delecyjnego nie zależy od wieku matki.
Czy 35 latka ma wyższe ryzyko zespołu delecyjnego niż 18 latka?
Nie, ryzyko zespołu delecyjnego u dziecka jest takie same bez względu na wiek matki. Oznacza to, że u 35-latki ryzyko zespołu delecyjnego nie będzie wyższe niż u kobiety w ciąży w wieku 18 lat.
Bibliografia
- Gillentine MA, Lupo PJ, Stankiewicz P, Schaaf CP. An estimation of the prevalence of genomic disorders using chromosomal microarray data. J Hum Genet. 2018 Jul;63(7):795-801. doi: 10.1038/s10038-018-0451-x. Epub 2018 Apr 24. PMID: 29691480; PMCID: PMC6019170.
- Levy B, Wapner R. Prenatal diagnosis by chromosomal microarray analysis. Fertil Steril. 2018 Feb;109(2):201-212. doi: 10.1016/j.fertnstert.2018.01.005. PMID: 29447663; PMCID: PMC5856154.
- Health Quality Ontario. Noninvasive Prenatal Testing for Trisomies 21, 18, and 13, Sex Chromosome Aneuploidies, and Microdeletions: A Health Technology Assessment. Ont Health Technol Assess Ser. 2019 Feb 19;19(4):1-166. PMID: 30847010; PMCID: PMC6395059.
- Srebniak MI, Joosten M, Knapen M, et al. Frequency of submicroscopic chromosomal aberrations in pregnancies without increased risk for structural chromosomal aberrations: systematic review and meta‐analysis. Ultrasound Obstetr Gynecol. 2018;51(4):445‐452.
- Rosenfeld JA, Patel A. Chromosomal Microarrays: Understanding Genetics of Neurodevelopmental Disorders and Congenital Anomalies. J Pediatr Genet. 2017 Mar;6(1):42-50. doi: 10.1055/s-0036-1584306. Epub 2016 May 30. PMID: 28180026; PMCID: PMC5288005.
- Jang W, Kim Y, Han E, Park J, Chae H, Kwon A, Choi H, Kim J, Son J, Lee S, Hong BY, Jang D, Han JY, Lee JH, Kim SY, Lee IG, Sung IK, Moon Y, Kim M, Park JH. Chromosomal Microarray Analysis as a First-Tier Clinical Diagnostic Test in Patients With Developmental Delay/Intellectual Disability, Autism Spectrum Disorders, and Multiple Congenital Anomalies: A Prospective Multicenter Study in Korea. Ann Lab Med 2019;39:299-310. https://doi.org/10.3343/alm.2019.39.3.299
- Vogels A, Fryns JP, Microdeletions and molecular genetics. Atlas of Genetics and Cytogenetics in Oncology and Haematology 2011. 10.4267/2042/38088.
- Srour M, Shevell M in Global Developmental Delay and Intellectual Disability (chapter 14), Editor(s): Rosenberg RN, Pascual JM, Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease (Fifth Edition), Academic Press, 2015: 151-161.
- Weise A, Mrasek K, Klein E, Mulatinho M, Llerena JC Jr, Hardekopf D, Pekova S, Bhatt S, Kosyakova N, Liehr T. Microdeletion and microduplication syndromes. J Histochem Cytochem. 2012 May;60(5):346-58. doi: 10.1369/0022155412440001. Epub 2012 Mar 6. PMID: 22396478; PMCID: PMC3351230.
- Wapner RJ, Martin CL, Levy B, Ballif BC, Eng CM, Zachary JM, Savage M, Platt LD, Saltzman D, Grobman WA, Klugman S, Scholl T, Simpson JL, McCall K, Aggarwal VS, Bunke B, Nahum O, Patel A, Lamb AN, Thom EA, Beaudet AL, Ledbetter DH, Shaffer LG, Jackson L. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med. 2012 Dec 6;367(23):2175-84. doi: 10.1056/NEJMoa1203382. PMID: 23215555; PMCID: PMC3549418.
- https://www.deciphergenomics. org
- https://www.orpha.net/consor/cgi-bin/index.php