Trisomia 21 - Zespół Downa

Spis treści
Zespół Downa — co to jest?
Zespół Downa (trisomia 21) jest to zespół charakterystycznych, choć w różnym stopniu wyrażonych i nasilonych wad spowodowany obecnością dodatkowego chromosomu 21 pary we wszystkich bądź w części komórek organizmu. W prawidłowych warunkach każda komórka ludzkiego ciała posiada 23 pary chromosomów. W przypadku zespołu Downa aberracja (nieprawidłowość) chromosomowa polega na tym, że pojawia się dodatkowy – trzeci chromosom 21. pary – stąd nazwa trisomia 21. Częstość występowania tego zespołu szacuje się na 1:650 – 1:700 żywo urodzonych dzieci. Średnio codziennie na świecie rodzi się ok. 700 dzieci z zespołem Downa, rocznie jest to ok. 255 tys.
Zespół Downa jako zespół pewnych cech fenotypowych został opisany w literaturze medycznej po raz pierwszy w 1866 roku przez angielskiego lekarza, który nazywał się John Langdon Down. To od jego nazwiska trisomia 21 została nazwana zespołem Downa. W 1959 roku francuski lekarz i genetyk Jerome LeJeune badając kariotypy osób z zespołem Downa odkrył, że jest on związany z trisomią chromosomu 21. Co ciekawe, praktycznie w tym samym czasie, bo 5 dni po J. LeJeune szkocka genetyk Patricia Jacobs opublikowała i tym samym potwierdziła to odkrycie.

Zespół Downa — przyczyny
Zespół Downa jest aberracją chromosomową, która polega na obecności dodatkowego chromosomu 21. pary. Oznacza to, że zamiast pary (dwóch) chromosomów 21. pary występują trzy chromosomy. Stąd też pochodzi druga nazwa tej choroby genetycznej – trisomia 21 lub trisomia 21. chromosomu.
Zespół Downa — dziedziczenie, rodzaje
Z punktu widzenia cytogenetyki zespół Downa może występować w 3 rodzajach:
- Prosta trisomia 21 – w tym przypadku każda komórka organizmu posiada dodatkowy (trzeci) chromosom 21, czyli innymi słowy zamiast dwóch posiada trzy kopie chromosomu 21. Prawidłowa komórka płciowa (komórka jajowa, plemnik) posiada 23 chromosomy (po jednym chromosomie z każdej pary). O tym jak przebiega proces powstawania komórek płciowych możesz przeczytać w artykule opisującym przebieg gametogenezy. (LINK do gametogenezy) Po połączeniu się żeńskiej i męskiej komórki płciowej (zapłodnienie) z prawidłową liczbą chromosomów powstanie zygota, która będzie posiadała pełen zestaw chromosomów, czyli 23 pary (w sumie 46 chromosomów). Przyczyną powstania tzw. prostej trisomii 21 jest nieprawidłowy rozdział chromosomów homologicznych (chromosomów tej samej pary) podczas podziału komórkowego czyli tzw. nondysjunkcja. W trisomii 21 najczęściej jest to nondysjunkcja podczas pierwszego podziału mejotycznego. (LINK do gametogeneza) Jej efektem jest powstanie gamet (komórek płciowych) – z nieprawidłową liczbą chromosomów. W przypadku zespołu Downa komórka jajowa lub plemnik zawierają 24 zamiast 23 chromosomów, a tym dodatkowym chromosomem jest chromosom 21. Innymi słowy – taka komórka płciowa zawiera dwa chromosomy 21 zamiast jednego. Jeżeli dojdzie do zapłodnienia np. komórki jajowej, która zawiera 24 chromosomy (a ten dodatkowy to drugi chromosom 21 i plemnika z prawidłową liczbą powstanie zygota, której garnitur chromosomowy to 46 +1 (czyli 23 pary chromosomów i dodatkowy, trzeci chromosom 21). Prosta trisomia 21 stanowi 95% wszystkich przypadków zespołu Downa.

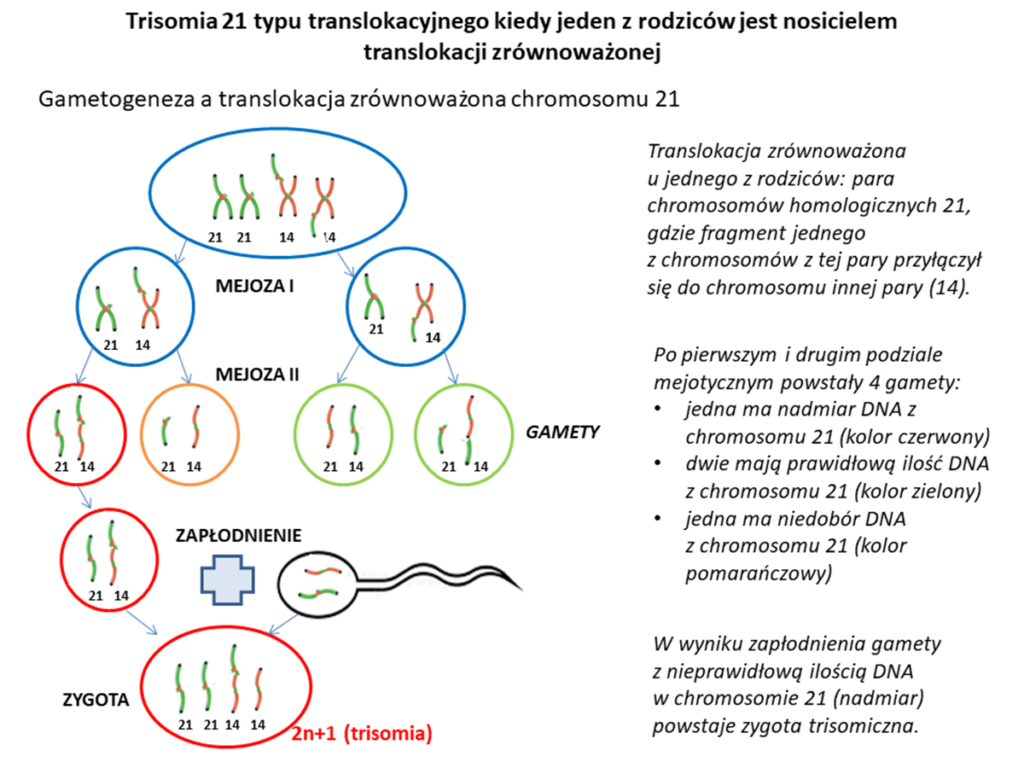
- Trisomia 21 typu translokacyjnego – ten rodzaj trisomii powstaje w wyniku translokacji, czyli przemieszczenia się fragmentu lub całego chromosomu i przyłączenia do chromosomu innej pary. Często jest to tzw. translokacja robertsonowska czyli taka, gdy dwa chromosomy akrocentryczne pękają w miejscu centromeru i krzyżowo łączą się ze sobą. Najczęściej występujący rodzaj zespołu Downa typu translokacyjnego to połączenie materiału genetycznego części chromosomu 21 z chromosomem 14, rzadziej z chromosomem 13,15 lub drugim chromosomem 21. Ten typ trisomii 21 występuje u ok. 2-4% osób dotkniętych tą wadą genetyczną. Translokacja może powstać w komórkach płciowych de novo, czyli przyłączenie się fragmentu chromosomu 21 do innego chromosomu nastąpi na etapie powstawania komórek jajowych lub plemników (ok. 75% wszystkich translokacji robertsonowskich w zespole Downa). Druga możliwa sytuacja to posiadanie translokacji zrównoważonej z zaangażowaniem chromosomu 21 przez jednego z rodziców. Translokacja zrównoważona to taki rodzaj translokacji, gdzie pomimo, że doszło do przemieszczenia się fragmentu chromosomu w inne miejsce, to materiał genetyczny nie został ani utracony, ani dodany. W takiej sytuacji pomimo, że fragment chromosomu 21 znajduje się w nieprawidłowym miejscu (np. jest połączony z ramieniem chromosomu 14), najczęściej nie wpływa to na ekspresję genów, ani na zdrowie osoby z taką translokacją. O takich osobach mówimy, że są nosicielami translokacji. Natomiast inaczej wygląda już sytuacja, jeżeli chodzi o potomstwo takiej osoby, ponieważ jej komórka płciowa może zawierać albo prawidłowy chromosom 14 albo chromosom 14 z przyłączonym fragmentem chromosomu 21, a do tego chromosom 21 – cały lub pozbawiony fragmentu, który przyłączył się do chromosomu 14. Jeżeli w komórce płciowej takiej osoby spotkają się ze sobą: prawidłowy chromosom 21 i chromosom 14 z przyłączonym fragmentem chromosomu 21 to mamy zdublowany materiał genetyczny znajdujący się na chromosomie 21, czyli tzw. translokację niezrównoważoną. Efektem zapłodnienia takiej komórki płciowej będzie zespół Downa. Ponieważ w tej sytuacji zespół Downa jest wynikiem odziedziczenia translokacji po jednym z rodziców, nazywany jest on rodzinnym. Takie przypadki to jedna trzecia wszystkich trisomii 21 typu translokacyjnego. Prawdopodobieństwo przekazania dziecku noszonej przez rodzica translokacji zależy od płci. Jeżeli nosicielem translokacji jest mężczyzna, to prawdopodobieństwo przekazania jej potomkowi wynosi ok. 3%, a jeżeli nosicielką jest kobieta to ok. 12%. Różnice te prawdopodobnie związane są z dużo większą selekcją i eliminacją plemników z nieprawidłowościami chromosomowymi.
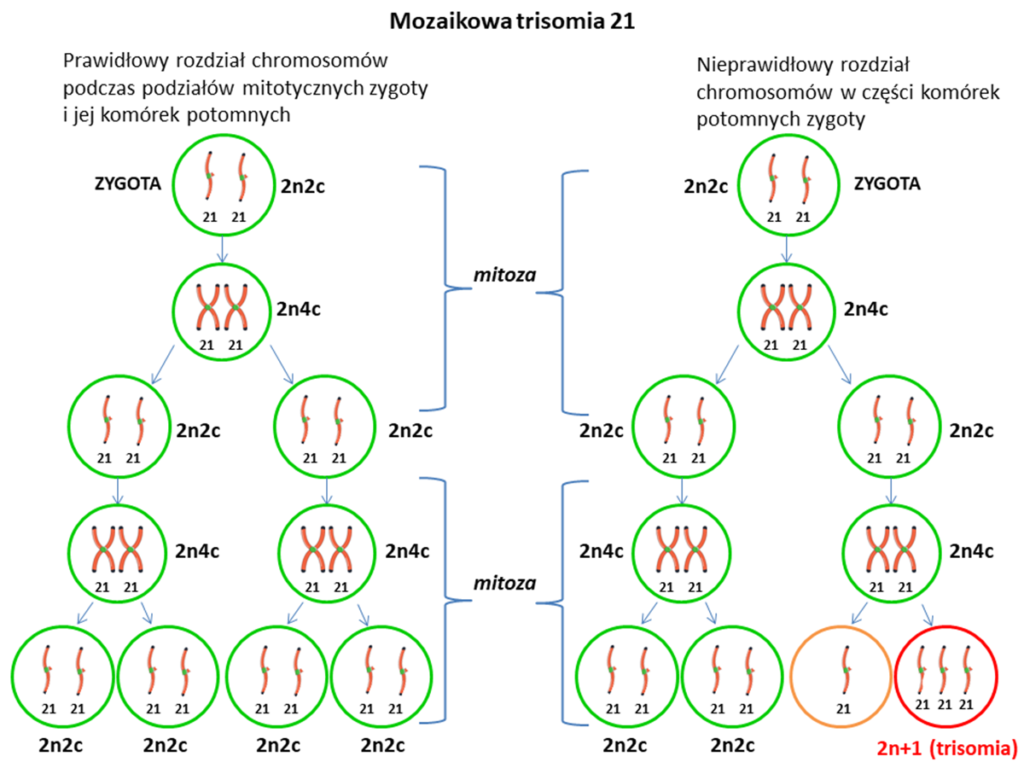
- Mozaikowa trisomia 21 – ten rodzaj zespołu Downa występuje najrzadziej (mniej niż 2% chorych) i często jest związana z tym, że cechy, które są typowe dla tej wady genetycznej są słabiej zarysowane, mniej wyraźne. Trisomia mozaikowa charakteryzuje się tym, że dodatkowy – trzeci chromosom 21 występuje nie we wszystkich, lecz tylko w części komórek organizmu. Taka sytuacja jest wynikiem nieprawidłowych podziałów komórkowych już po zapłodnieniu.
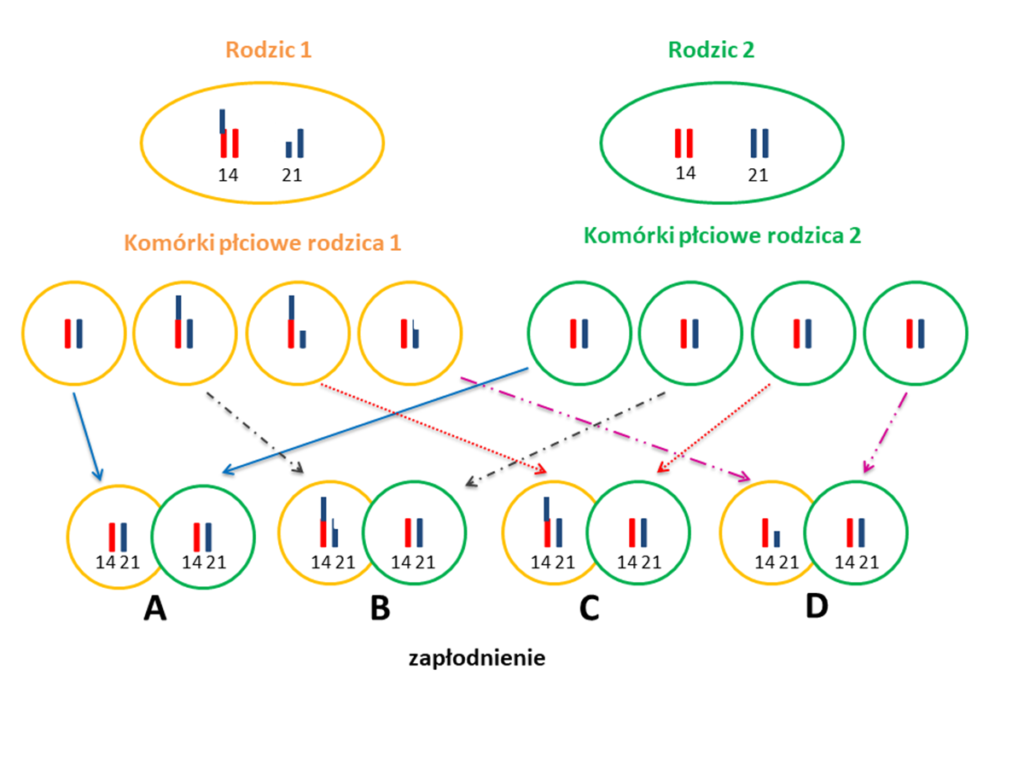
Czy rodzic, który posiada translokację zrównoważoną zawsze przekaże ją swojemu potomstwu, a dziecko będzie miało zespół Downa?
Niekoniecznie, tak jak pisaliśmy wcześniej, ok. 30% dzieci, których jedno z rodziców ma translokację zrównoważoną jest obarczone trisomią 21. W sytuacji gdy u rodzica występuje translokacja zrównoważona 21. chromosomu istnieje kilka możliwości dla każdej z ciąż:
- (A) Dziecko może odziedziczyć prawidłowy garnitur chromosomów i nie będzie obciążone aberracją chromosomową związaną z chromosomem 21.
- (B) Dziecko może odziedziczyć dokładnie taką samą translokację zrównoważoną jaką ma rodzic. W takiej sytuacji w większości przypadków u dziecka nie ma problemów zdrowotnych będących wynikiem takiej translokacji.
- (C) Dziecko może odziedziczyć translokację niezrównoważoną, której wynikiem będzie zespół wad genetycznych związanych z trisomią 21.
- (D) Ciąża zakończy się poronieniem.
Poniższy schemat pokazuje możliwe opcje układu chromosomów 14. i 21. w komórkach płciowych rodziców, gdzie: rodzic 1 ma translokację zrównoważoną (fragment chromosomu 21 jest połączony z chromosomem 14), a rodzic 2 ma prawidłowy układ chromosomów i możliwe opcje ich połączenia podczas zapłodnienia (literami A, B, C, D pokazane są schematycznie opisane powyżej opcje).
Chromosom 21
Chromosom 21 jest najmniejszym chromosomem w całym garniturze chromosomowym człowieka pod względem ilości par zasad (46,7 mln), jego DNA ma stosunkowo niską „gęstość” genową w porównaniu z pozostałym chromosomami. Na chromosomie 21 znajduje się ok. 2% genomu, w tym 236 genów kodujących. Dla porównania, na chromosomach 18 i 13, których trisomie są drugą i trzecią najczęściej występującą aneuploidią po trisomii 21, znajdują się odpowiednio 269 i 322 geny kodujące, a najwięcej genów kodujących, bo aż 2056, posiada chromosom 1 (Esemble genome browser release, 2021). W zależności od rodzaju trisomii 21, stopnia rearanżacji chromosomu 21 (np. w translokacjach) obraz fenotypowy osoby z zespołem Downa będzie nieco inny. Każda z osób chorujących na trisomię 21. chromosomu ma swój własny obraz kliniczny choroby. Są pewne cechy wspólne, związane chociażby z wyglądem zewnętrznym, czy też częstością niektórych wad i schorzeń towarzyszących u osób z zespołem Downa, lecz nawet ich nasilenie różni się między osobami.
Poniżej prezentujemy wybrane geny chromosomu 21, których potrojenie, mutacja, delecja itd. mogą przyczyniać się do określonych dysfunkcji i objawów zespołu Downa:
- geny APP (ang.amyloid precursor protein) i BACE2 (ang. beta secretase 2)– potrojenie tych genów związane jest z wczesnym rozwojem choroby Alzheimera (najnowsze dane naukowe pokazują, że potrojenie genu APP zwiększa ryzyko wczesnego rozwoju choroby Alzheimera również u osób zdrowych, tzn. bez trisomii)
- gen DSCR1 (ang. Down syndrome critical region 1) – jego potrojona obecność odpowiada za zespół cech dymorficznych, wadę serca oraz rozwój choroby Alzheimera
- geny: DYRK1A, SIM2 (ang. single-minded homolog 2) i SYNJ1 (ang. synaptojanin 1) – związane z problemami z uczeniem się i z zapamiętywaniem
- gen RCAN1 (ang. regulator of calcineurin 1) – jeden z genów odpowiedzialnych za obniżenie odpowiedzi immunologicznej
- gen ITSN1 (intersectin-1) – upośledzenie umysłowe
- gen KCNE 1 (ang. potassium voltage-gated channel subfamili E member 1) – upośledzenie właściwości elektrofizjologicznych mięśnia sercowego
- gen RUNX1 (ang. runt-related transcription factor 1) – mutacje genu prowadzą do rodzinnej małopłytkowości, a także rozwoju białaczek, u osób z delecją genu występuje rozszczep podniebienia
- geny: Olig1 (ang. oligodendrocyte transcription factor 1) i Olig2 (ang. oligodendrocyte lineage transcription factor 2) – potrojenie tych genów wiąże się z upośledzeniem rozwoju mózgu, tworzeniu się synaps, zaburzeniami zachowania
- gen TTC3 (ang. tetratricopeptide repeat domain–containing protein 3) – niedobór białka, które koduje ten gen może być powiązany z zaburzeniami rozwoju poznawczego oraz budowy mózgu
- gen C21orf 29 (ang. chromosome 21 open reading frame 29 peptide), druga nazwa tego genu to TSPEAR (ang. thrombospondin-type lamining domain and ear repeats) – gen padaczki
- geny COL6A1 (ang. collagen, type VI, alpha-1) , COL6A2 (ang. collagen, type VI,alpha-2), – zaburzone różnicowanie i rozwój komórek, które może powodować choroby nerwowo-mięśniowe, wady serca i naczyń krwionośnych, wady stawów, kośćca itp.
- gen COL18A1 (ang. collagen, type XVIII, alpha-1) – krótkowzroczność, postępująca degeneracja ciała szklistego i siatkówki, odwarstwienia siatkówki

Czynniki ryzyka urodzenia dziecka z zespołem Downa:
- wiek matki – ryzyko wystąpienia zespołu Downa u potomstwa rośnie wraz z wiekiem matki
wiek matki (w latach) | ryzyko wystąpienia zespołu Downa u dziecka |
20 | 1/1600 |
25 | 1/1300 |
30 | 1/1000 |
35 | 1/365 |
40 | 1/90 |
45 | 1/30 |
- translokacja zrównoważona u jednego z rodziców
- wcześniejsze urodzenie dziecka z zespołem Downa (ryzyko urodzenia drugiego dziecka z trisomią 21 wzrasta o ok. 1%)
Objawy trisomii 21 (objawy zespołu Downa)
U osób z trisomią 21. chromosomu występują specyficzne cechy dysmorficzne, czyli pewne zmiany w wyglądzie zewnętrznym będące wynikiem tej aberracji chromosomowej. Pojedyncze cechy dysmorficzne mogą występować również u ludzi zdrowych czy też osób z innymi wadami genetycznymi. Niemniej jednak w przypadku zespołu Downa zestaw tych cech jest bardzo charakterystyczny i rozpoznawalny. Nie oznacza to, że wszystkie osoby z trisomią 21 wyglądają tak samo. Każda z nich ma swoje indywidualne cechy wyglądu, swój kolor oczu, kolor i kształt brwi, kolor włosów, wyraz twarzy czy też uśmiech.
Do najczęściej występujących, charakterystycznych cech wyglądu osób z zespołem Downa należą:
- umiarkowane małogłowie, spłaszczenie głowy w części potylicznej, krótkogłowie (skrócenie głowy w wymiarze przednio-tylnym)
- płaski i okrągły profil twarzy
- nisko osadzone i szeroko rozstawione oczy
- zmarszczka nakątna (Fałd skórny przykrywający przyśrodkowy kąt oka)
- skośnie ustawione szpary powiekowe
- kąciki ust skierowane ku dołowi
- jasne plamki na tęczówce oka – odbarwienia tęczówek (tzw. plamek Brushfielda)
- mały, krótki nos z płaską nasadą i szerokim grzbietem
- nisko osadzone, małe małżowiny uszne, często mały, nieobecny lub nieprawidłowo ukształtowany płatek ucha
- wąskie i mocno wysklepione podniebienia twarde (tzw. gotyckie podniebienie)
- wydatne usta, czasem wywinięta warga dolna, usta często otwarte
- duży, wystający język o nierównej powierzchni, utrudniający zamknięcie ust
- księżycowate, nieprawidłowo rozstawione zęby
- krótka szyja z fałdem skórnym na karku
- małe, szerokie dłonie z pojedynczą bruzdą zgięciową (tzw. małpia bruzda, czyli gruba, pojedyncza bruzda biegnąca w poprzek wewnętrznej części dłoni w miejscu gdzie większość ludzi ma dwie płytsze linie główne – bruzdy)
- krótki palec mały u ręki
- szeroka przerwa między paluchem a drugim palcem stopy (tzw. bruzda sandałowa)
- miękkie, delikatne, często rzadkie włosy skóry głowy
- sucha, szorstka, marmurkowata skóra
- niski wzrost

Ponadto u noworodków występują takie cechy charakterystyczne jak:
- hipotonia mięśniowa (obniżone napięcie mięśni)
- osłabiony odruch Moro (inaczej odruch obejmowania, jest to pierwotny odruch noworodków, które w reakcji na określony bodziec energicznie odrzucają rączki w bok, naprężają się, prostują nóżki, wyginają plecy w łuk i odchylają główkę do tyłu)
- nadmierna ruchomość w stawach
Osoby z zespołem Downa odznaczają się dość niskim wzrostem, osiągając zwykle ok. 150 cm. Obserwuje się za to tendencję do nadmiernego przyrostu masy ciała, co tłumaczy się wolniejszym tempem metabolizmu.
Wady i schorzenia towarzyszące zespołowi Downa
Wady wrodzone serca u osób z zespołem Downa
Występują u ok. 50% urodzonych dzieci z trisomią 21. Do niedawna były one najczęstszą przyczyną śmierci dzieci z tą wadą genetyczną w ciągu pierwszych 2 lat życia. Obecnie, dzięki rozwojowi kardiochirurgii dziecięcej wiele z tych wad może być skutecznie operowanych.
- najczęściej występującą wadą (ok. 40%) jest ubytek w przegrodzie międzyprzedsionkowej serca
- drugą co do częstości występowania wadą jest ubytek w przegrodzie międzykomorowej serca (32%),
- 10% osób z zespołem Downa rodzi się ze wspólnym kanałem przedsionkowo-komorowym, u 6% występuje tetralogia Fallota (sinicza wada serca obejmuje cztery nieprawidłowości anatomiczne: ubytek w przegrodzie międzykomorowej, zwężenie drogi odpływu z prawej komory, przemieszczenie aorty na prawo, nad przegrodę międzykomorową oraz przerost prawej komory) lub drożny przewód tętniczy (przetrwały, otwarty przewód Botalla)
- u ok. 30% osób z trisomią 21. chromosomu występuje więcej niż jedna wrodzona wada serca

Wady przewodu pokarmowego u osób z trisomią 21
Występują u ok. 5% osób z zespołem Downa i stanowią drugą po wadach serca przyczynę interwencji chirurgicznych u osób z trisomią 21.
Do najczęstszych należą:
- zwężenie lub zarośnięcie odbytu lub dwunastnicy
- przerostowe zwężenie odźwiernika
- choroba Hirschsprunga (wrodzone nieprawidłowe unerwienia jelita, spowodowane brakiem śródściennych zwojów nerwowych w dystalnym odcinku jelita grubego, co sprawia, że ten odcinek jelita jest w stanie permanentnego skurczu i przyczynia się do niedrożności jelita)
- wąskie i wiotkie podniebienie, gruby, sztywny język oraz wady uzębienia i zgryzu, które utrudniają życie i połykanie pokarmu – oprócz tego, że są same w sobie wadą pierwszego odcinka układu pokarmowego, to mogą przyczyniać się do kolejnych zaburzeń – np. problemy z trawieniem i wchłanianiem, czy też problemy z motoryką żołądka i jelit
Oprócz wrodzonych wad układu pokarmowego, u osób z zespołem Downa obserwuje się wyższe ryzyko wystąpienia takich schorzeń i dolegliwości jak:
- celiakia
- refluks żołądkowo-przełykowy
- przewlekłe zaparcia
- okresowe biegunki
Zaburzenia neurologiczne i zaburzenia narządu ruchu u osób z zespołem Downa
- cechą charakterystyczną praktycznie wszystkich dzieci z trisomią 21 jest hipotonia mięśniowa, która powoduje wolniejszy rozwój motoryczny dzieci (obniżone napięcie mięśniowe wpływa na nadmierną ruchomość i wiotkość stawów, co z kolei skutkuje niestabilnością postawy ciała i chodu, niepewnością w trakcie chodzenia, biegania itp. czy też gorszą koordynacją ruchową, dlatego dzieci z zespołem Downa muszą wkładać więcej energii w każdą aktywność fizyczną, ruch)
- większa skłonność do urazów jak zwichnięcia w związku z wiotkością aparatu więzadłowego stawów i złamania kości w wyniku obniżonej gęstości masy kostnej, co osłabia strukturę kości (dodatkowo niekorzystnie na gęstość masy kostnej wpływa niska aktywność fizyczna, która u osób z trisomią 21 jest często spowodowana trudnościami motorycznymi wynikającymi z obniżonego napięcia mięśni)
- u 13% dzieci z zespołem Downa stwierdza się niestabilność kręgu szczytowego i obrotnika (pierwszy i drugi kręg szyi), rozpoznanie tej nieprawidłowości jest szczególnie istotne w kontekście planowania ćwiczeń i aktywności fizycznej, która ma wspierać rozwój psychoruchowy dzieci
- osoby z zespołem Downa są bardziej narażone na demencję (po 50. r.ż. ryzyko wzrasta do 70%) i wcześniejszy rozwój choroby Alzheimera
- trisomia 21. chromosomu wiąże się z wyższym ryzykiem rozwoju padaczki, występuje również korelacja rozwoju padaczki z chorobą Alzheimera (50% osób z trisomią 21, które będą borykać się z chorobą Alzheimera dotkną również ataki padaczki)
- u prawie wszystkich osób z zespołem Downa występują zaburzenia poznawcze, problemy z uczeniem się związane z niepełnosprawnością intelektualną o różnym stopniu nasilenia: w większości jest to niepełnosprawna intelektualnie w stopniu lekkim (iloraz inteligencji 50-70) lub w stopniu umiarkowanym (iloraz inteligencji 35-50)
- u osób z trisomią 21 możemy też zaobserwować wolniejszy rozwój mowy i problemy logopedyczne, takie jak mało wyraźna mowa, trudności w wymowie niektórych głosek, zaburzenia fonetyczne czy niski poziom rozumienia mowy
- częściej niż w ogólnej populacji występują zaburzenia zachowania, zaburzenia psychiczne oraz autyzm (ok. 7%)

Zaburzenia endokrynologiczne i metaboliczne u osób z trisomią 21
- choroby tarczycy to najczęstszy problem endokrynologiczny osób z zespołem Downa, częstość występowania niedoczynności tarczycy jest 28-35 razy wyższa niż dla ogółu populacji (1 na 113-141 osób), u 13-34% osób wykrywa się podwyższony poziom przeciwciał tarczycowych; rzadziej stwierdza się nadczynność tarczycy
- cukrzyca typu 1 – dotyka osoby z trisomią 21 trzykrotnie częściej niż wynosi średnia zachorowalność dla całej populacji
- otyłość – w porównaniu z ogółem populacji, u osób z zespołem Downa otyłość występuje dwa do czterech razy częściej (33-71% chorych), duży skok BMI obserwuje się już w ok. 3 r.ż. u dziewczynek i w 5 r.ż. u chłopców
- dyslipidemia (zaburzenia stężenia lipidów i lipoprotein we krwi) dotyczy ok. 58% osób z trisomią
- hiperleptynemia i leptynooporność – pojawiają się badania wskazujące na to, że na chromosomie 21 znajdują się również geny związane z większą predyspozycją do tych zaburzeń, co z kolei niekorzystnie wpływa na ilość tkanki tłuszczowej oraz jest wysokim czynnikiem ryzyka zespołu metabolicznego, chorób układu krążenia, cukrzycy typu 2 itp.
Zaburzenia układu moczowo-płciowego u chorych z zespołem Downa
- opóźnione dojrzewanie u obu płci
- u dziewczynek najczęściej jest to opóźnione adrenarche (faza rozwojowa, w której następuje zwiększone wydzielanie androgenów przez korę nadnerczy – dzieje się to w okresie poprzedzającym okres dojrzewania płciowego bądź na wczesnym etapie dojrzewania płciowego, u dziewczynek ok. 7 r.z.) oraz opóźniona pierwsza miesiączka
- u chłopców może występować wnętrostwo, mikropenis, dysgenezja jąder (zaburzenia różnicowania i rozwoju jąder), wodniaki jąder, niska objętość nasienia, ubogie owłosienie pachowe i łonowe; niektóre z wymienionym zaburzeń mogą być przyczyną męskiej niepłodności
Choroby zębów i przyzębia w zespole Downa
- opóźnione i nietypowe wyrzynanie się zębów
- wrodzony brak zębów (agenezja)
- zęby o nieprawidłowym kształcie
- choroby szkliwa
- wady zgryzu
- próchnica
Choroby i wady narządów zmysłów u osób z trisomią 21
- narząd wzroku: wady refrakcji (wady wzroku) i astygmatyzm (60-70%), anomalie tęczówki (38-90%), zez (32-34%), zaćma (25-85%), niedowidzenie (amblyopia, 10-26%), oczopląs (5-30%), stożek rogówki (5-8%), zapalenie powiek (2-7%)
- narząd słuchu: niedosłuch (ok. połowa osób z trisomią 21 ma problemy ze słuchem), który często jest wynikiem patologii ucha środkowego (m.in. malformacje dotyczące trąbki słuchowej Eustachiusza, zwężenie przewodu słuchowego zewnętrznego oraz hipoplazja wyrostka sutkowego), co może powodować ostre zapalenia ucha środkowego, przewlekłe wysiękowe zapalenia ucha środkowego czy też perforację błony bębenkowej ; do wysiękowego zapalenia ucha środkowego dodatkowo predysponuje częsty u osób z trisomią 21 przerost migdałka gardłowego i migdałków podniebiennych

Zaburzenia i choroby hematologiczne a zespół Downa
U noworodków z zespołem Downa często stwierdza się łagodne zaburzenia hematologiczne, tzw. HANDS (ang. hematological abnormalities in a newborn with Down syndrome), które najczęściej mijają w pierwszych 3 tygodniach życia.
Należą do nich:
- neutrofilia, która występuje u 80% noworodków z trisomią 21 (zwiększona liczba granulocytów obojętnochłonnych czyli neutrofili będących najliczniejszą grupą wśród białych krwinek i wchodzącą w skład układu odpornościowego)
- trombocytopenia dotyka 66% urodzonych dzieci z zespołem Downa (małopłytkowość, obniżona liczba płytek krwi)
- policytemia, którą obserwuje się u 34% noworodków z trisomią 21. chromosomu (inaczej nadkrwistość, czerwienica, czyli zwiększona liczba erytrocytów, czasem towarzyszy jej wzrost liczby białych krwinek i płytek krwi).
U ok. 10% noworodków z zespołem Downa stwierdza się przemijający zespół mieloproliferacyjny (ang. TMD, transient myeloproliferative disorder. Najczęściej jest on diagnozowany w pierwszym tygodniu życia dziecka i u ok. 80% dzieci ulega samoistnemu wycofaniu (remisji) w ciągu 3 miesięcy. TMD charakteryzuje się występowaniem zwiększonej liczby białych krwinek we krwi dziecka, a w tym kilkanaście do kilkudziesięciu procent stanowią komórki zwane blastami (są to komórki prekursorowe, z których w szpiku powstają białe krwinki). Do objawów sugerujących występowanie przemijającego zespołu mieloproliferacyjnego należą między innymi powiększenie wątroby i śledziony, zmiany w obrębie serca, trzustki, przedłużająca się żółtaczka oraz zajęcie szpiku kostnego. Czasem może również wystąpić łagodna niedokrwistość oraz trombocytopenia.
Osoby z zespołem Downa są szczególnie predysponowane (10-krotnie wyższe ryzyko) do występowania białaczki. Ok. 2% wszystkich ostrych białaczek limfoblastycznych (ALL, ang. acute lymphoblastic leukemia) wieku dziecięcego i ok. 10% ostrych białaczek szpikowych (AML, ang. acute myeloid leukemia) wieku dziecięcego rozpoznaje się u dzieci z trisomią 21. Osoby, u których rozpoznano przemijający zespół mieloproliferacyjny po urodzeniu są w grupie zwiększonego ryzyka wystąpienia ostrej białaczki szpikowej lub zespołu mielodysplastycznego w przyszłości.
Podatność na infekcje układu oddechowego osób z zespołem Downa
- zwiększona skłonność do infekcji dróg oddechowych jest związana zarówno z dysfunkcjami układu odpornościowego, jak i innymi czynnikami (można je nazwać czynnikami nieimmunologicznymi)
- do czynników nieimmunologicznych należą m.in. anomalie w budowie anatomicznej ucha wewnętrznego (opisane wyżej) i dróg oddechowych (dotyczą ok. 75% dzieci z trisomią 21) jak np. laryngomalacja i tracheomalacja (wrodzona wiotkość krtani i tchawicy związane z nieprawidłowym wykształceniu chrząstek je budujących), krtań o kształcie lejkowatym, hipoplazja płuc oraz słabo rozwinięte mięśnie międzyżebrowe, obniżone napięcie mięśniowe, wrodzone wady serca; innymi czynnikami przyczyniającymi się do częstszych infekcji są obturacyjny bezdech senny i refluks żołądkowo-przełykowy
- osłabienie funkcji układu odpornościowego może wiązać się z: zaburzeniami parametrów i funkcji makrofagów, monocytów, limfocytów B i T, komórek NK, stężenia przeciwciał IgA, IgM, IgG
- u dzieci z zespołem Downa obserwuje się nie tylko wyższą zachorowalność na zakażenia układu oddechowego w porównaniu z dziećmi o prawidłowym genotypie, lecz również dłuższy czas trwania infekcji i silniejsze natężenie objawów
Obturacyjny bezdech senny – dotyczy on 30-80% osób z zespołem Downa, u których występuje szereg czynników predysponujących do bezdechów. Należą do nich m.in. hipoplazja żuchwy, duży, wystający język, anomalie krtaniowo-tchawicze, przerost migdałka gardłowego i migdałków podniebiennych oraz otyłość. Nieleczony bezdech senny może prowadzić do wielu powikłań układu sercowo-naczyniowego, zaburzeń neurobehawioralnych, rozwoju stanów zapalnych, większej podatności na infekcje, czy też pośrednio prowadzić do otyłości i zespołu metabolicznego oraz może być przyczyną moczenia nocnego.
Wyjątkowe i mocne strony osób z zespołem Downa:
- inteligencja praktyczna, rozwiązywanie konkretnych problemów, a słabą myślenie abstrakcyjne, matematyczne.
- rozwój psychospołeczny chorych, zdolność nawiązywania relacji, dojrzałość społeczna i emocjonalna są zwykle na dobrym poziomie
Diagnostyka zespołu Downa
Na etapie I trymestru ciąży wykonywane są nieinwazyjne badania prenatalne, dzięki którym lekarz ginekolog określa ryzyko wystąpienia trisomii 21. chromosomu (oraz trisomii 18 i 13). W tym celu wykonuje się:
- USG prenatalne pierwszego trymestru ciąży, które wykonywane jest pomiędzy 11. a 13. tygodniem ciąży i ocenia się w nim m.in. takie parametry jak przezierność karku (NT) oraz kość nosowa (NB) (USG PRENATALNE PIERWSZEGO TRYMESTRU)
- test podwójny (tzw. test PAPP-A) – badanie biochemiczne wykonywane na próbce krwi pobranej od kobiety ciężarnej, gdzie oznacza się poziom dwóch substancji: białka ciążowego A (tzw. białka PAPP-A) oraz wolnej podjednostki beta-hCG, czyli hormonu wydzielanego przez łożysko (optymalnym momentem do pobrania krwi do badań biochemicznych I trymestru jest wg Polskiego Towarzystwa Ginekologicznego 10 – 11 tydzień ciąży) (TEST PODWÓJNY CZYLI TEST PAPPA-A)

Na podstawie uzyskanych wyników (NT, NB, wolna podjednostka β-hCG i białko PAPP-A) oraz uwzględniając wiek kobiety ciężarnej lekarz przy wykorzystaniu specjalnego algorytmu może określić jakie jest prawdopodobieństwo urodzenia dziecka z zespołem Downa. Warto podkreślić, że tylko wspólna analiza wszystkich tych parametrów pozwala na uzyskanie wiarygodnych wyników oceny ryzyka trisomii 21. Według FMF (ang. Fetal Medicine Foundation) analiza skuteczności wymienionych powyżej nieinwazyjnych metod screeningowych pokazuje, że w przypadku gdy weźmiemy pod uwagę wszystkie wymienione wskaźniki (tzw. test zintegrowany) możemy wykryć 95% ciąż z zespołem Downa (przy 5% odsetku wyników fałszywie pozytywnych). Skuteczność tych wskaźników obniża się, jeśli rozpatrujemy je oddzielnie lub w połączeniu, ale nie wszystkie równocześnie. Jeśli chcesz zobaczyć jak kształtują się współczynniki wykrywalności w zależności od wykorzystanych parmaterów badań nieinwazyjnych – przeczytaj artykuł TEST PODWÓJNY CZYLI TEST PAPP-A.
Warto również dodać, że w Polsce u kobiet w ciąży test PAPP-A nie jest obowiązkowy. Istnieje możliwość refundacji tego testu dla kobiet w ciąży będących powyżej 35. r.ż.. Według opinii Polskiego Towarzystwa Ginekologicznego badania prenatalne powinny być proponowane do wykonania wszystkim kobietom ciężarnym, bez względu na wiek. Pomimo, że wg statystyk ryzyko urodzenia dziecka z trisomią 21 rośnie po 35. r.ż., to więcej dzieci z zespołem Downa rodzą mamy, które nie ukończyły 35 lat. Są to proste prawa matematyki – generalnie więcej młodszych kobiet rodzi dzieci, niż tych po 35 r.ż.. Stąd też, pomimo, że statystycznie ryzyko urodzenia dziecka z trisomią 21. chromosomu u tych kobiet jest niższe, to ogólna liczba dzieci z tą nieprawidłowością chromosomową urodzonych przez młodsze mamy jest duża w porównaniu ze znacznie mniejszą całkowitą liczbą dzieci urodzonych przez matki w wieku 35+.
Wyniki nieinwazyjnych testów prenatalnych pierwszego trymestru dają nam informację jakie jest ryzyko urodzenia dziecka z trisomią 21, ale na ich podstawie nie stawia się diagnozy. Za niskie ryzyko urodzenia dziecka z zespołem Downa, zgodnie z rekomendacjami Zespołu ekspertów Polskiego Towarzystwa Ginekologicznego oraz Polskiego Towarzystwa Genetyki Człowieka (2017), uważa się ryzyko, które określone w teście zintegrowanym wynosi < 1:1000.
W diagnostyce prenatalnej trisomii 21 wykorzystuje się zarówno badania inwazyjne jaki i nieinwazyjne. Optymalnie jeżeli w pierwszej kolejności wykorzystujemy te możliwości diagnostyki, które nie niosą ryzyka dla diagnozowanego maleństwa. Dlatego w pewnych zakresach ryzyka proponuje się alternatywne drogi, które pozwalają uzyskać rodzicom dodatkowe informacje, zanim podejmą decyzję o badaniach inwazyjnych.
Przykładowo w sytuacji gdy ryzyko wyliczone na podstawie wyniku badania przesiewowego I trymestru ciąży wynosi pomiędzy 1:100 a 1:1000 eksperci zalecają wykonanie dodatkowych nieinwazyjnych badań prenatalnych jakimi są przesiewowe badania genetyczne wykonanego na wolnym, płodowym DNA, tzw. testy NIPT, które są dodatkowym narzędziem określającym prawdopodobieństwo urodzenia potomstwa z trisomią 21. (CO TO JEST NIPT) Natomiast jeżeli ryzyko wyliczone w teście zintegrowanym wynosi > 1:100, zalecenia sugerują już pominąć badania nieinwazyjne typu NIPT ( np: Nifty, Nifty Pro, Harmony, Panorama, Sanco, Nace, Neobona, Veracity, Veragene ) i wykonać inwazyjne prenatalne badania diagnostyczne, co nie znaczy, że rodzic nie może zdecydować inaczej.(INFORMACJE OGÓLNE O INWAZYJNYCH BADANIACH PRENATALNYCH) Są to badania, które analizują materiał genetyczny komórek płodu, np. amniopunkcję (AMINOPUNKCJA). Celem takiego badania jest określenie kariotypu płodu (KARIOTYP), czyli jakościowa i ilościowa ocena zestawu chromosomów jakie ma dziecko. Tylko na tej podstawie możemy postawić właściwą diagnozę. Coraz częściej właśnie wykonuje się ocenę kariotypu płodu jeszcze przed urodzeniem, z materiału pobranego w trakcie inwazyjnych badań prenatalnych.

Po urodzeniu wstępne rozpoznanie jest formułowane na podstawie cech zewnętrznego wyglądu dziecka oraz badania przedmiotowego (np. ocena odruchów, ocena napięcia mięśniowego). Ostateczne potwierdzenie diagnozy uzyskuje się na podstawie wspomnianych już badań cytogenetycznych określających kariotyp dziecka. Badanie wykonuje się poprzez pobranie próbki krwi dziecka (bardzo rzadko również innych tkanek) – albo jeszcze w szpitalu zaraz po urodzeniu, albo w poradni genetycznej, do której dziecko otrzymuje skierowanie.
Kariotyp podawany jest w formie zapisu:
- w przypadku prawidłowego kariotypu zapis dla odpowiednio chłopca i dziewczynki wygląda następująco: 46XY lub 46XX
- w przypadku potwierdzenia zespołu Downa zapis kariotypu dla odpowiednio chłopca i dziewczynki wygląda następująco 47XY,+21 lub 47XX,+21, co oznacza dodatkowy chromosom (zamiast 46, jest ich 47) z zaznaczeniem, który z chromosomów jest nadmiarowy (+21 – czyli chromosom 21)
Dodatkowo wykonanie badania kariotypy pozwala na określenie ryzyka ponownego wystąpienia zespołu Downa w kolejnych ciążach. Najczęściej to ryzyko jest niskie i wynosi ok. 1%, ale wszystko zależy od typu trisomii 21 i kariotypu rodziców, o czym pisaliśmy wyżej w części dotyczącej podłoża genetycznego trisomii 21.
Po urodzeniu dziecko z zespołem Downa wymaga również diagnostyki specjalistycznej – przede wszystkim kardiologicznej, aby wykluczyć czy też ewentualnie potwierdzić wady serca. Bardzo istotne jest też rozpoznanie i leczenie chorób tarczycy, gdyż może to mieć bardzo istotny wpływ na dalszy rozwój dziecka. Często potrzebna jest wczesna rehabilitacja, a także wsparcie innych specjalistów w zależności od potrzeb zdrowotnych dziecka.
Leczenie zespołu Downa
Niestety w przypadku choroby genetycznej jaką jest trisomia 21 nie ma możliwości całkowitego wyleczenia, ani też leczenia przyczynowego. Opiekę nad osobą chorą sprawuje lekarz POZ (pediatra, lekarz rodzinny, lekarz internista). Poza tym osoba z zespołem Downa wymaga wielospecjalistycznej opieki medycznej, której zakres zależy od stanu zdrowia danej osoby. Często potrzebne jest wsparcie chociażby okulisty, neurologa, endokrynologa, diabetologa, fizjoterapeuty czy też logopedy. Prowadzone leczenie jest leczeniem objawowym, a w przypadku np. wad serca czy przewodu pokarmowego często jest niezbędna interwencja chirurgiczna. Ważny jest stały monitoring zdrowia osoby z trisomią 21, badania przesiewowe i diagnostyczne, które pozwolą na jak najszybsze wykrycie ewentualnych nieprawidłowości jak np. zaburzenia słuchu i wzroku, niedobory immunologiczne czy też choroby hematologiczne. Leczenia ma na celu niwelowanie objawów tak, aby jak najbardziej poprawić komfort życia osoby chorej i jej opiekunów. Istotna jest ciągła ocena rozwoju fizycznego, intelektualnego dziecka. Zwraca się również uwagę na bardzo ważną rolę profilaktyki otyłości.
Rokowanie trisomii 21
Dzięki postępom medycyny średnia długość życia osób z zespołem Downa znacząco się wydłużyła. Na początku XX wieku chorzy z trisomią 21 przeżywali średnio 9 lat, w 1960 roku była to już 30 lat, a obecnie średnia życia osób z zespołem Downa wynosi 56 lat. Oczywiście jest ona uzależniona od wielu czynników zdrowotnych, jak chociażby od obecności wrodzonych wad serca, przewodu pokarmowego czy zaburzeń endokrynologicznych.
Dzieci trisomią 21 już od pierwszych lat życia wymagają stałej opieki rehabilitacyjnej oraz intelektualnej, mogą potrzebować pomocy ze strony logopedy czy też pedagoga oraz częstych wizyt u innych specjalistów. Bardzo często osoby z łagodnym stopniem upośledzenia dobrze radzą sobie w dorosłym życiu i nie potrzebują bardzo dużego zaangażowania ze strony opiekunów, niejednokrotnie mogą również prowadzić życie zawodowe. Z kolei przy cięższych stopniach upośledzenia osoba z zespołem Downa może potrzebować pomocy w codziennych czynnościach, jak ubieranie się, mycie, zakupy, gotowanie czy załatwianie formalności w urzędach.
Bibliografia
- Akhtar F, Bokhari SRA. Down Syndrome. [Updated 2021 Mar 30]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK526016/
- Moreau M, Benhaddou S, Dard R, Tolu S, Hamzé R, Vialard F, Movassat J, Janel N. Metabolic Diseases and Down Syndrome: How Are They Linked Together? Biomedicines. 2021 Feb 22;9(2):221. doi: 10.3390/biomedicines9020221. PMID: 33671490; PMCID: PMC7926648.
- Altuna M, Giménez S, Fortea J. Epilepsy in Down Syndrome: A Highly Prevalent Comorbidity. J Clin Med. 2021 Jun 24;10(13):2776. doi: 10.3390/jcm10132776. PMID: 34202784; PMCID: PMC8268854.
- Kevin L Howe, Premanand Achuthan, James Allen, Jamie Allen, Jorge Alvarez-Jarreta, M Ridwan Amode, Irina M Armean, Andrey G Azov, Ruth Bennett, Jyothish Bhai, Konstantinos Billis, Sanjay Boddu, Mehrnaz Charkhchi, Carla Cummins, Luca Da Rin Fioretto, Claire Davidson, Kamalkumar Dodiya, Bilal El Houdaigui, Reham Fatima, Astrid Gall, Carlos Garcia Giron, Tiago Grego, Cristina Guijarro-Clarke, Leanne Haggerty, Anmol Hemrom, Thibaut Hourlier, Osagie G Izuogu, Thomas Juettemann, Vinay Kaikala, Mike Kay, Ilias Lavidas, Tuan Le, Diana Lemos, Jose Gonzalez Martinez, José Carlos Marugán, Thomas Maurel, Aoife C McMahon, Shamika Mohanan, Benjamin Moore, Matthieu Muffato, Denye N Oheh, Dimitrios Paraschas, Anne Parker, Andrew Parton, Irina Prosovetskaia, Manoj P Sakthivel, Ahamed I Abdul Salam, Bianca M Schmitt, Helen Schuilenburg, Dan Sheppard, Emily Steed, Michal Szpak, Marek Szuba, Kieron Taylor, Anja Thormann, Glen Threadgold, Brandon Walts, Andrea Winterbottom, Marc Chakiachvili, Ameya Chaubal, Nishadi De Silva, Bethany Flint, Adam Frankish, Sarah E Hunt, Garth R IIsley, Nick Langridge, Jane E Loveland, Fergal J Martin, Jonathan M Mudge, Joanella Morales, Emily Perry, Magali Ruffier, John Tate, David Thybert, Stephen J Trevanion, Fiona Cunningham, Andrew D Yates, Daniel R Zerbino, Paul Flicek, Ensembl 2021, Nucleic Acids Research, Volume 49, Issue D1, 8 January 2021, Pages D884–D891.
- Amr NH. Thyroid Disorders in Subjects with Down Syndrome: An Update. Acta Biomed. 2018 Mar 27;89(1):132-139. doi: 10.23750/abm.v89i1.7120. PMID: 29633736; PMCID: PMC6357620.
- Whooten R, Schmitt J, Schwartz A. Endocrine manifestations of Down syndrome. Curr Opin Endocrinol Diabetes Obes. 2018 Feb;25(1):61-66. doi: 10.1097/MED.0000000000000382. PMID: 29135488; PMCID: PMC6382276.
- Satgé D, Seidel MG. The Pattern of Malignancies in Down Syndrome and Its Potential Context With the Immune System. Front Immunol. 2018 Dec 19;9:3058. doi: 10.3389/fimmu.2018.03058. PMID: 30631328; PMCID: PMC6315194.
- Patiroglu T, Cansever M, Bektas F. Underlying factors of recurrent infections in patients with down syndrome. North Clin Istanb. 2018 Jan 29;5(2):163-168. doi: 10.14744/nci.2017.69379. PMID: 30374487; PMCID: PMC6191560.
- Hithersay R, Hamburg S, Knight B, Strydom A. Cognitive decline and dementia in Down syndrome. Curr Opin Psychiatry. 2017 Mar;30(2):102-107. doi: 10.1097/YCO.0000000000000307. PMID: 28009725.
- de la Piedra MJ, Alberti G, Cerda J, Cárdenas A, Paul MA, Lizama M. Alta frecuencia de dislipidemias en niños y adolescentes con Síndrome de Down [High frequency of dyslipidemia in children and adolescents with Down Syndrome]. Rev Chil Pediatr. 2017;88(5):595-601. Spanish. doi: 10.4067/S0370-41062017000500004. PMID: 29546943.
- Kazemi M, Salehi M, Kheirollahi M. Down Syndrome: Current Status, Challenges and Future Perspectives. Int J Mol Cell Med. 2016 Summer;5(3):125-133. Epub 2016 Aug 10. PMID: 27942498; PMCID: PMC5125364.
- Buonuomo PS, Bartuli A, Mastrogiorgio G, Vittucci A, Di Camillo C, Bianchi S, Pires Marafon D, Villani A, Valentini D. Lipid profiles in a large cohort of Italian children with Down syndrome. Eur J Med Genet. 2016 Aug;59(8):392-5. doi: 10.1016/j.ejmg.2016.06.005. Epub 2016 Jun 23. PMID: 27343989.
- Asim A, Kumar A, Muthuswamy S, Jain S, Agarwal S. „Down syndrome: an insight of the disease”. J Biomed Sci. 2015 Jun 11;22(1):41. doi: 10.1186/s12929-015-0138-y. PMID: 26062604; PMCID: PMC4464633.
- Ram G, Chinen J. Infections and immunodeficiency in Down syndrome. Clin Exp Immunol. 2011 Apr;164(1):9-16. doi: 10.1111/j.1365-2249.2011.04335.x. Epub 2011 Feb 24. PMID: 21352207; PMCID: PMC3074212.
- Chakrabarti L, Best TK, Cramer NP, Carney RS, Isaac JT, Galdzicki Z, Haydar TF. Olig1 and Olig2 triplication causes developmental brain defects in Down syndrome. Nat Neurosci. 2010 Aug;13(8):927-34. doi: 10.1038/nn.2600. Epub 2010 Jul 18. PMID: 20639873; PMCID: PMC3249618.
- Mégarbané A, Ravel A, Mircher C, Sturtz F, Grattau Y, Rethoré MO, Delabar JM, Mobley WC. The 50th anniversary of the discovery of trisomy 21: the past, present, and future of research and treatment of Down syndrome. Genet Med. 2009 Sep;11(9):611-6. doi: 10.1097/GIM.0b013e3181b2e34c. PMID: 19636252.
- Starbuck JM. On the antiquity of trisomy 21: moving towards a quantitative diagnosis of Down syndrome in historic material culture. Journal of Contemporary Anthropology. 2011;2:18–44.
- Alexander K. C. Leung, MD, Alexander K. C. Leung, MD, W. Lane M. Robson, MD, Hardally R. Hegde, MD. Translocation Down (Trisomy 21) Syndrome. Consultant for Pediatricians 2006 Aug, 5(8).
- American Academy of Pediatrics. Committee on Genetics. American Academy of Pediatrics: Health supervision for children with Down syndrome. Pediatrics. 2001 Feb;107(2):442-9. doi: 10.1542/peds.107.2.442. PMID: 11158488.
- Gamis AS, Smith FO. Transient myeloproliferative disorder in children with Down syndrome: clarity to this enigmatic disorder. Br J Haematol. 2012 Nov;159(3):277-87. doi: 10.1111/bjh.12041. Epub 2012 Sep 12. PMID: 22966823.
- Gamis AS, Alonzo TA, Gerbing RB, Hilden JM, Sorrell AD, Sharma M, Loew TW, Arceci RJ, Barnard D, Doyle J, Massey G, Perentesis J, Ravindranath Y, Taub J, Smith FO. Natural history of transient myeloproliferative disorder clinically diagnosed in Down syndrome neonates: a report from the Children’s Oncology Group Study A2971. Blood. 2011 Dec 22;118(26):6752-9; quiz 6996. doi: 10.1182/blood-2011-04-350017. Epub 2011 Aug 17. PMID: 21849481; PMCID: PMC3245202.
- Hattori M, Fujiyama A, Taylor TD, Watanabe H, Yada T, Park HS, Toyoda A, Ishii K, Totoki Y, Choi DK, Groner Y, Soeda E, Ohki M, Takagi T, Sakaki Y, Taudien S, Blechschmidt K, Polley A, Menzel U, Delabar J, Kumpf K, Lehmann R, Patterson D, Reichwald K, Rump A, Schillhabel M, Schudy A, Zimmermann W, Rosenthal A, Kudoh J, Schibuya K, Kawasaki K, Asakawa S, Shintani A, Sasaki T, Nagamine K, Mitsuyama S, Antonarakis SE, Minoshima S, Shimizu N, Nordsiek G, Hornischer K, Brant P, Scharfe M, Schon O, Desario A, Reichelt J, Kauer G, Blocker H, Ramser J, Beck A, Klages S, Hennig S, Riesselmann L, Dagand E, Haaf T, Wehrmeyer S, Borzym K, Gardiner K, Nizetic D, Francis F, Lehrach H, Reinhardt R, Yaspo ML; Chromosome 21 mapping and sequencing consortium. The DNA sequence of human chromosome 21. Nature. 2000 May 18;405(6784):311-9. doi: 10.1038/35012518. Erratum in: Nature 2000 Sep 7;407(6800):110. PMID: 10830953.
- Rekomendacje Zespołu Ekspertów Polskiego Towarzystwa Ginekologicznego oraz Polskiego Towarzystwa Genetyki Człowieka w zakresie przesiewowego badania genetycznego wykonanego na wolnym płodowym DNA, Ginekologia i Perinatologia Praktyczna 2017, tom 2, nr 5, 230–233.
- Rekomendacje Sekcji Ultrasonografii Polskiego Towarzystwa Ginekologów i Położników w zakresie przesiewowej diagnostyki ultrasonograficznej w ciąży o przebiegu prawidłowym – 2020 r.