Zespół Smith-Magenis’a

Spis treści
Zespół Smith-Magenis’a – informacje ogólne
Zespół Smith-Magenis’a (Delecja 17p11.2) po raz pierwszy opisano w 1982 r. To rzadka choroba o podłożu genetycznych charakteryzująca się m.in. dysmorfią twarzy, opóźnieniem rozwoju oraz upośledzeniem umysłowym. Częstotliwość występowania szacuje się na 1:25000, choroba może wystąpić u obu płci.
Podstawy genetyczne zespołu Smith-Magenis’a
Zespół Smith-Magenis’a należy do grupy chorób związanych z aberracjami chromosomalnymi. To jeden z tzw. zespołów przyległego genu.
Istotą tej choroby w większości przypadków jest delecja fragmentu krótkiego ramienia chromosomu 17 w regionie 11 podregionie 2. W zależności od dokładnej wielkości utraconego fragmentu DNA zespół Smith-Magenis’a może dawać różne objawy o różnym nasileniu.
Wiele z genów obecnych we wspomnianym regionie p11.2 chromosomu 17 nie została do tej pory zidentyfikowana, dlatego nie jest możliwe jednoznaczne powiązanie konkretnych symptomów z utratą konkretnego genu. Najprawdopodobniej za wiele objawów odpowiada delecja genu RAI1 (z ang. Retinoic Acid Induced 1).
W większości przypadków zespół Smith-Magenis’a nie jest dziedziczony, a delecja powstaje de novo na wczesnym etapie rozwoju prenatalnego.
Zespół Smith-Magenis’a – objawy
Najbardziej charakterystycznym elementem obrazu klinicznego zespołu Smith-Magenis’a są:
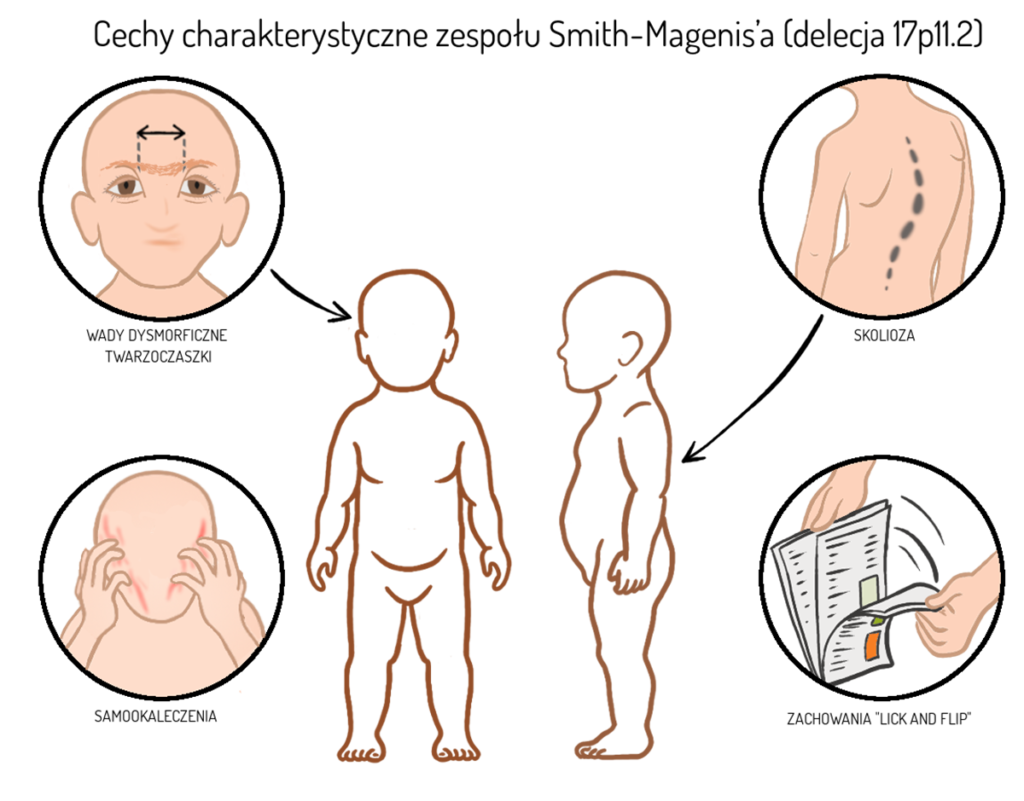
- samookaleczenia (np. zrywanie paznokci z palców dłoni i stóp, uderzenia głową o ścianę, gryzienie, drapanie, wyrywanie włosów)
- tzw. zachowania „lick and flip”, czyli bardzo szybkie przerzucanie stron książki, zeszytu czy gazety, połączone ze ślinieniem palca i całkowitym brakiem zainteresowania treścią
- samoprzytulanie
Dodatkowo pacjenci często cierpią z powodu zaburzeń snu, które prawdopodobnie są spowodowane nieprawidłowym rytmem wydzielania melatoniny.
Inne symptomy zespołu Smith-Magenis’a to:
- Wady dysmorficzne twarzoczaszki – m.in. niedorozwój środkowej części twarzy, wysokie czoło, krótki, zadarty nos, nienaturalnie mała żuchwa, szerokie, najczęściej otwarte usta, opadające kąciki, wysunięty język. Wady te pogłębiają się wraz z wiekiem
- Nieprawidłowe przybieranie na masie ciała związane z trudnościami z przyjmowaniem pokarmów z powodu m.in. zmniejszeniem napięcia i ruchliwości mięśni języka, nieprawidłowym uzębieniem, refluksem
- Skolioza, występująca średnio u ok. 60% pacjentów i pogłębiająca się wraz z wiekiem
- Deformacje tęczówki oka, zez, krótkowzroczność
- Częste zapalenia ucha środkowego i zatok
- Chrapliwy, nosowy głos
- Upośledzenie umysłowe w stopniu od umiarkowanego do znacznego
- Zaburzenia pamięci, koncentracji, problemy z uczeniem się
- Opóźnione nabywanie umiejętności samodzielnego siadania, chodzenia
- Zaburzenia behawioralne np. napady złości, agresywność, moczenie nocne, nieposłuszeństwo, próby ciągłego zwracania na siebie uwagi
Leczenie zespołu Smith-Magenis’a
Postępowanie terapeutyczne u pacjentów z zespołem Smith-Magenis’a obejmuje wyłącznie leczenie objawowe. Jednym z najczęstszych elementów jest stosowanie leków psychotropowych w celu poprawy zachowania, zdolności koncentracji i obniżenia częstotliwości epizodów samookaleczania. Zarówno dzieci, jak również całe rodziny powinny być objęte opieką psychologiczną.
Rokowania zespołu Smith-Magenis’a
Dane dotyczące długości życia osób z rozpoznanym zespołem Smith-Magenis’a są ograniczone, jednak są przypadki, kiedy pacjenci żyli ponad 80 lat.