Trisomia 18 - Zespół Edwardsa

Spis treści
Zespól Edwardsa
Zespół Edwardsa (trisomia 18) jest to zespół wad spowodowany obecnością dodatkowego chromosomu 18 lub dodatkowego fragmentu chromosomu 18. Jest drugą najczęściej występującą trisomią autosomalną. Częstość występowania ocenia się na 1:6000 – 1:8000 żywo urodzonych dzieci, natomiast w związku z tym, że większość przypadków trisomii 18 prowadzi do samoistnego poronienia, całkowita częstotliwość tego zespołu wad genetycznych jest szacowana na 1:2500 – 1:2600 ciąż. Częstotliwość żywych urodzeń jest wyższa w przypadku dziewczynek z zespołem Edwardsa i wynosi ok. 60%
Po raz pierwszy opis zespołu wielu wad wrodzonych związanych z trisomią 18 pojawił się w czasopiśmie naukowym Lancet w 1960 r. Jego autorem był John Hilton Edwards, brytyjski lekarz i genetyk medyczny. W tym samym roku, kilka miesięcy później zespół Edwardsa został opisany przez amerykański zespół lekarzy i naukowców (Smith D.W. et al.).

Podłoże genetyczne zespołu Edwardsa
Zespół Edwardsa jest nieprawidłowością chromosomową, która związana jest z obecnością dodatkowego, „nadprogramowego” materiału genetycznego chromosomu 18. Prawidłowy zestaw chromosomów w komórkach człowieka to 23. pary chromosomów homologicznych. Zespół Edwardsa charakteryzuje obecność dodatkowego całego (zamiast dwóch, występują trzy chromosomy 18.) lub części chromosomu 18. pary.
Cytogenetycznie trisomię 18 możemy podzielić na:
- Całkowita trisomia 18 – każda komórka organizmu posiada pełny dodatkowy (trzeci) chromosom 18. Ten rodzaj trisomii 18. chromosomu występuje najczęściej i dotyczy ok. 94% przypadków. Przyczyną całkowitej trisomii 18 jest nondysjunkcja chromosomów, czyli nieprawidłowy rozdział chromosomów homologicznych (chromosomów tej samej pary) w trakcie podziałów komórkowych gametogenezy. Odpowiedni rozdział chromosomów homologicznych, a następnie rozdział chromatyd siostrzanych, który ma miejsce podczas powstawania gamet żeńskich i męskich warunkuje powstanie komórki jajowej i plemników z prawidłowym zestawem chromosomów (każda gameta ma 23 chromosomy, czyli po 1 chromosomie z każdej pary). Dzięki temu w wyniku zapłodnienia powstanie zygota posiadająca prawidłowy garnitur chromosomowy, czyli zestaw 46 chromosomów (23 pary). W wyniku błędu podczas rozdziału materiału genetycznego może powstać gameta, w której ilość chromosomów jest nieprawidłowa. W tym przypadku dotyczy to 18. pary chromosomów, kiedy to gameta zamiast jednego, ma dwa chromosomy 18. W takim przypadku, po zapłodnieniu zygota będzie miała nie dwa, a trzy chromosomy 18 (stąd nazwa trisomia 18), a co za tym idzie w sumie 47 chromosomów (46 + 1 dodatkowy). Co ciekawe, całkowita trisomia 18 jest najczęściej wynikiem nondysjunkcji podczas drugiego podziału mejotycznego (GAMETOGENEZA), w przeciwieństwie do innych ludzkich trisomii, które są głównie wynikiem błędu w rozdziale chromosomów podczas pierwszego podziału mejotycznego. Najczęściej dodatkowy chromosom 18. jest pochodzenia matczynego, co prawdopodobnie wynika z wczesnej selekcji i eliminacji plemników z nieprawidłowościami chromosomowymi.

- Trisomia 18 typu mozaikowego – charakteryzuje się obecnością w organizmie osoby z zespołem Edwardsa populacji komórek z prawidłowym zestawem chromosomów (23 pary, czyli 46 chromosomów) oraz komórek z nieprawidłowym zestawem chromosomów (46 + 1), w których występuje całkowita trisomia 18. Typ mozaikowy zespołu Edwardsa dotyczy niespełna 5% przypadków. Najczęściej jest on wynikiem nieprawidłowych podziałów mitotycznych (GAMETOGENEZA) części komórek, które mają miejsce już po zapłodnieniu. Fenotyp osób z mozaikowym typem trisomii 18 może być skrajnie zróżnicowany, od bardzo trudnych zdrowotnie przypadków kończących się wczesną śmiercią, do zdrowych, fenotypowo „normalnych” dorosłych, u których ten rodzaj trisomii stwierdza się dopiero po badaniach genetycznych wykonanych w ramach diagnostyki po urodzeniu dziecka z trisomią 18. Warto również podkreślić, że nie ma żadnej korelacji pomiędzy rodzajem i natężeniem objawów zespołu Edwardsa, a procentowym udziałem komórek z mozaicyzmem we krwi obwodowej czy też w fibroblastach skóry .
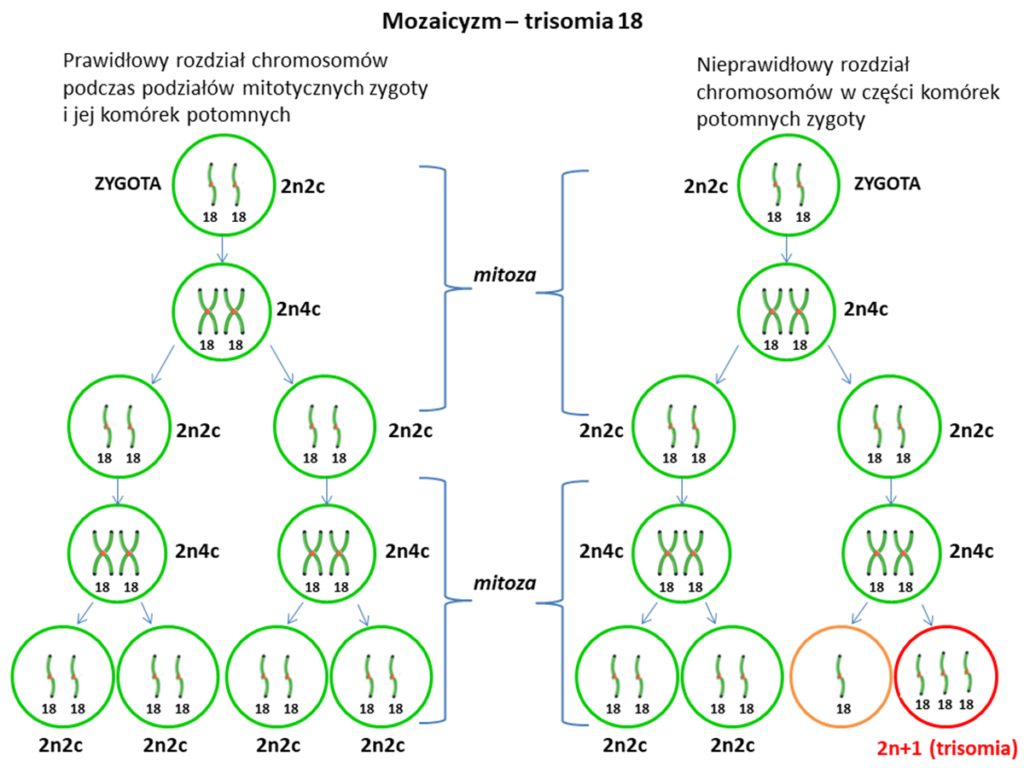
- Częściowa trisomia 18 – w komórkach znajdują się 2 prawidłowe chromosomy 18 oraz dodatkowo fragment chromosomu 18 (zazwyczaj jest to długie ramię chromosomu 18 – czyli długie ramię występuje w trzech kopiach). Ten rodzaj trisomii 18 najczęściej jest wynikiem obecności translokacji zrównoważonej występującej u jednego z rodziców. Translokacja zrównoważona związana jest z pęknięciem chromosomu i przemieszczeniem się jego fragmentu w inne miejsce (może to być np. drugi chromosom homologiczny tej samej pary bądź inny chromosom z całego zestawu chromosomów). W translokacji zrównoważonej zasadniczo nie zmienia się ilość materiału genetycznego, ale następuje zmiana jego rozmieszczenia w genomie. Dzięki temu u osoby, która jest nosicielem translokacji zrównoważonej, taka aberracja najczęściej nie przejawia się fenotypowo, natomiast może mieć już wpływ na zdrowie potomstwa osoby z taką translokacją. Nosiciel translokacji zrównoważonej może mieć potomstwo, u którego stwierdza się translokację niezrównoważoną. Jeżeli fragment chromosomu 18. przyłączy się do innego chromosomu, to może powstać komórka płciowa, która ma ubytek lub nadmiar DNA. Taka komórka płciowa (np. komórka jajowa) po połączeniu z drugą gametą, w tym przypadku plemnikiem da zygotę z nieprawidłowym zestawem chromosomów (ubytek bądź nadmiar DNA). Innymi słowy efektem zapłodnienia takiej komórki jajowej może być zespół Edwardsa. Translokacja niezrównoważona, która w efekcie da zespół Edwardsa może również powstać de novo, np. w wyniku pęknięcia dwóch niehomologicznych chromosomów bądź przypadkowej rekombinacji niehomologicznej w mejozie.
Częściowa trisomia 18 występuje najrzadziej i dotyczy max. 2% przypadków zespołu Edwardsa. Fenotyp osób z częściową trisomią 18 jest bardzo zróżnicowany, co jest związane z tym jaki obszar chromosomu 18. jest trisomiczny.

Czy rodzic posiadający translokację zrównoważoną zawsze urodzi potomstwo z translokacją niezrównoważoną, której efektem będzie zespół Edwardsa?
Nie, nie zawsze. Ryzyko zespołu Edwardsa u potomstwa rodziców, z których jedno ma translokację zrównoważoną z zaangażowaniem chromosomu 18 wynosi do 30%. Jakie są możliwości?
A. Dziecko może odziedziczyć całkowicie prawidłowy układ chromosomów i nie będzie miało trisomii 18.
B. Dziecko może odziedziczyć taką samą zmianę chromosomową jaką ma jego rodzic, czyli w tym przypadku translokacje zrównoważoną, co również nie będzie powodowało trisomii 18.
C. Dziecko może urodzić się z translokacją niezrównoważoną, która da zespół Edwardsa.
D. Ciąża zakończy się poronieniem.
Poniższy przykładowy schemat pokazuje możliwe opcje układu chromosomów 16. i 18. w komórkach płciowych rodziców, gdzie: rodzic 1 ma translokację zrównoważoną (fragment chromosomu 18 jest połączony z chromosomem 16), a rodzic 2 ma prawidłowy układ chromosomów i możliwe opcje ich połączenia podczas zapłodnienia (litery A, B, C, D odpowiadają konsekwencjom zapłodnienia w zależności od opcji połączenia się komórek płciowych, co zostało opisane powyżej).

Chromosom 18
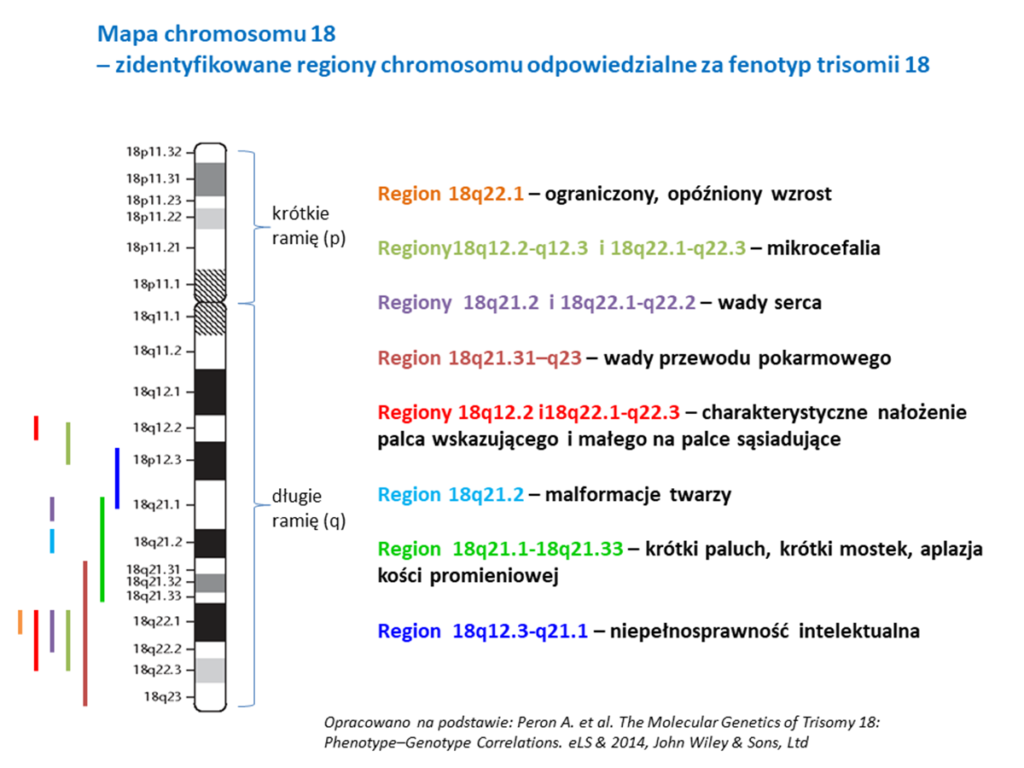
Chromosom 18 jest chromosomem submetacentrycznym (jego centromer położony jest w pobliżu środka chromosomu, ale nie jest dokładnie w środku, co sprawia, że podczas metafazy i anafazy podziału komórkowego (GAMETOGENEZA) przybiera kształt litery L. DNA tego chromosomu liczy ok. 80 milionów par zasad i stanowi ok. 2,7% ludzkiego materiału genetycznego znajdującego się w komórce. Jest jednym z mniejszych chromosomów, dla porównania największy – chromosom 1 ma prawie 249 mln par zasad. Ale z kolei w porównaniu z chromosomem 21 jest dwukrotnie dłuższy (chromosom 21 ma 48,7 mln par zasad). Liczba genów kodujących chromosomu 18 szacowana jest na 269 i jest 7,5-krotnie mniejsza od liczby genów chromosomu 1 (2058 genów kodujących), natomiast porównywalna do liczby genów kodujących chromosomu 21, który ma ich 236 (Esemble genome browser release, 2021). Badania genetyczne wskazują na to, że główny wpływ na fenotyp zespołu Edwardsa ma trisomia długiego ramienia chromosomu 18, natomiast jego krótkie ramię nie pełni znaczącej roli w tej chorobie genetycznej. Ponieważ trisomia 18 jest rzadką chorobą genetyczną (pomimo, że to druga co do częstości występowania trisomia), cały czas trwają badania, których celem jest identyfikacja regionów i genów chromosomu 18, które pełnią kluczową rolę w zespole Edwardsa. W literaturze najczęściej wymieniane są 2 regiony krytyczne, oba zlokalizowane są na długim ramieniu chromosomu 18: 18q12.1-18q21.2 oraz 18q22.3-qter. Zidentyfikowano również kilka regionów, których trisomia została powiązana z różnymi cechami klinicznymi zespołu Edwardsa. Zostały one przedstawione na poniższej grafice.
Czynniki ryzyka urodzenia dziecka z zespołem Edwardsa:
- Ryzyko urodzenia dziecka z dodatkowym, 18 chromosomem wzrasta wraz z wiekiem matki (np. u kobiet w 35 roku życia jest dwukrotnie wyższe niż u kobiet w 30 roku życia, a z kolei w 40 roku życia w porównaniu do 30 roku życia to ryzyko wzrasta prawie dziesięciokrotnie).
W poniższej tabeli zebrano dane dotyczące związku między wiekiem matki, a ryzykiem urodzenia dziecka z jakąkolwiek aneuploidią.
- Statystyki pokazują, że trisomia 18. chromosomu dotyczy czterokrotnie częściej dziewczynek niż chłopców.
- Ryzyko ponownego urodzenia dziecka z trisomią 18 wynosi ok. 1-1,5%.
- Czynnikiem ryzyka jest również translokacja zrównoważona chromosomu 18 u rodzica (prawdopodobieństwo wynosi ok. 30%).
Objawy trisomii 18
Zespół Edwardsa jest trisomią, która ma bardzo zróżnicowany obraz kliniczny. Oznacza to, że zarówno na etapie pre-, jak i postnatalnym objawy trisomii 18 nie są takie same u wszystkich dzieci z zespołem Edwardsa.
Do charakterystycznych objawów zespołu Edwardsa na etapie prenatalnym, które mogą, choć nie muszą być widoczne w badaniu USG na poszczególnych etapach ciąży należą m.in.:
- mała aktywność ruchowa płodu
- pojedyncza tętnica pępowinowa w sznurze pępowinowym (prawidłowo jest parzysta)
- przedurodzeniowy niedobór wzrastania i niska masa ciała płodu w stosunku do wieku ciąży
- charakterystyczne ułożenie dłoni w zaciśniętą piąstkę tak, że palec wskazujący i piąty (mały) są nałożone na pozostałe palce
- wady serca
- niedrożność przełyku
- głowa „truskawkowa” (czaszka o kształcie truskawki)
- przepuklina przeponowa
- przepuklina sznura pępowinowego
- rozszczep kręgosłupa
- agenezja ciała modzelowatego
- rozszczepy twarzy
- stopy szpotawe
- torbiele sznura pępowinowego
Dzieci z zespołem Edwardsa, które przyszły na świat najczęściej mają niską masę urodzeniową oraz szereg cech dysmorficznych i wad narządowych oraz układowych. Dotychczas w literaturze możemy znaleźć ponad 125 anomalii, które są powiązane z trisomią 18. chromosomu. Dotyczą one m.in. nieprawidłowości struktur kostno-stawowych (twarzoczaszki, kręgosłupa, kończyn), mózgowia, serca, układu oddechowego, pokarmowego oraz moczowo-płciowego. Poszczególne osoby z trisomią 18 różnią się między sobą zarówno spektrum anomalii i wad, które je dotykają, jak i stopniem ich nasilenia. Niemniej jednak liczne wady budowy pociągają za sobą nieprawidłowości w funkcjonowaniu organizmu i uniemożliwiają prawidłowy rozwój.

Wady czaszki i twarzy w trisomii 18
- dolichocefalia (inaczej długogłowie) – wada rozwojowa czaszki, która charakteryzuje się długą i wąską głową spłaszczoną po bokach)
- mikrocefalia (inaczej małogłowie) – wada rozwojowa czaszki, która charakteryzuje się nienaturalnie małymi rozmiarami czaszki, a co za tym idzie – mniejszą masą mózgowia)
- poszerzenie szwów głowowych i ciemiączek
- wypukła potylica
- trójkątna, asymetryczna twarz
- mikrognacja (hipoplazja) żuchwy – wada, która charakteryzuje się cofnięciem górnej szczęki w stosunku do żuchwy, co objawia się ustawieniem dolnych zębów przed górnymi
- mikroftalmia (inaczej małoocze) – w okresie życia płodowego gałka oczna nie osiąga prawidłowych rozmiarów i ostatecznie jest mniejsza niż być powinna w określonym wieku dziecka
- hiperteloryzm – duża odległość pomiędzy gałkami ocznymi
- wąskie szpary powiekowe
- fałd skórny obecny w okolicy kąta przyśrodkowego oka (tzw. zmarszczka nakątna)
- opadanie górnych powiek
- wydatne grzbiety nadoczodołowe
- nisko osadzone, małe i zniekształcone małżowiny uszne z niezawiniętym obrąbkiem
- obecność wrodzonych przydatków przedusznych
- mikrostomia – małe usta, znaczne ograniczenia i trudności w otwieraniu jamy ustnej
- wąskie łuki podniebienne
- rozszczep wargi i podniebienia
- nadmiar skóry na szyi
Wady układu kostno-stawowego w zespole Edwardsa
- krótka szyja
- krótki mostek
- lejkowata klatka piersiowa
- duża odległość pomiędzy małymi brodawkami sutkowymi (bądź brak brodawek)
- niepełne skostnienie obojczyka
- wady rozwojowe kręgów
- skolioza
- wąska miednica
- zwichnięcie stawu biodrowego
- ograniczenie ruchomości stawów biodrowych w zakresie odwodzenia
- brak kości promieniowej (jedna z dwóch kości przedramienia)
- artrogrypoza – wrodzona sztywność stawów
- specyficzne ułożenie palców dłoni podczas zaciskania pięści – palec wskazujący nachodzi na palec środkowy, a palec mały (piąty) nachodzi na palec serdeczny (czwarty)
- wady anatomiczne palców – zniekształcone, przykurczowo ustawione palce u rąk (kampodaktylia), słabo rozwinięte kciuki, krótki paluch u stopy, syndaktylia (palcozrost), niedorozwój paznokci
- stopa końsko-szpotawa (złożona deformacja stóp)
- wystająca kość piętowa (tzw. stopa cepowata)
Wady wrodzone serca
Występują u ok. 90% urodzonych dzieci z zespołem Edwardsa.:
- najczęstsze wady to ubytek przegrody międzykomorowej i przetrwały przewód tętniczy
- tetralogia Fallota (sinicza wada serca, która obejmuje cztery nieprawidłowości anatomiczne: ubytek w przegrodzie międzykomorowej, zwężenie drogi odpływu z prawej komory, przemieszczenie aorty na prawo, nad przegrodę międzykomorową oraz przerost prawej komory)
- wielozastawkowe wady serca
- dwuujściowa (dwuodpływowa) prawa komora serca (DORV, ang. double outlet right ventricle) oraz grupa wad serca spowodowanych nieprawidłowym połączeniem górnych i dolnych poduszeczek wsierdziowych – występują rzadziej, u ok. 10% noworodków z trisomią 18 i najczęściej są przyczyną wczesnej śmierci
Wady układu oddechowego u osób z trisomią 18
- hipoplazja płuc – wada wrodzona związana z niewykształceniem (różnego stopnia) elementów naczyniowych, oskrzelowych i miąższowych tkanki płucnej
- tracheobronchomalacja – wrodzona wiotkość tchawicy i oskrzeli (ściany dróg oddechowych są słabe i mogą ulec zwężeniu lub zapadnięciu)
- laryngomalacja – wrodzona wiotkość krtani
- wady przepony
Wady przewodu pokarmowego u osób z zespołem Edwardsa
- przepukliny – najczęściej pachwinowa lub pępkowa
- atrezja przełyku (wrodzona niedrożność przełyku) z towarzyszącą jej przetoką tchawiczo-przełykową
- wrodzone przerostowe zwężenie odźwiernika (pylorostenoza)
- wrodzona atrezja (niedrożność) jelita krętego
- malrotacja jelit (niedokonany zwrot jelit), która może prowadzić do niedrożności jelit
- uchyłek Meckela (pozostałość po przewodzie żółtkowo-jelitowym, który jest obecny w życiu płodowym)
Wady układu nerwowego
Dotyczą ok. 30% osób z trisomią 18.
Należą do nich:
- hipoplazja móżdżku
- przepuklina oponowo-mózgowa
- sekwencja malformacyjna: akrania (bezczaszkowie, czyli częściowy lub całkowity brak kości czaszki) – egencefalia (stan, w którym z powodu braku jamy czaszki, mózgowie znajduje się poza czaszką, występuje dezorganizacja tkanki mózgowej) – anencefalia (bezmózgowie, czyli szczątkowy rozwój mózgowia
- wodogłowie
- holoprozencefalia – związane jest z niedokonanym podziałem przodomózgowia na dwie półkule, któremu często towarzyszą malformacje środkowej części twarzy
- zespół Arnolda Chiariego (malformacja Arnolda-Chiariego) – przemieszczenia się struktur tyłomózgowia do kanału, w którym przebiega rdzeń kręgowy, co skutkuje wystąpieniem objawów neurologicznych, spowodowanych uciskiem wywieranym na rdzeń przez nieprawidłowo ułożoną część mózgu
- hipoplazja ciała modzelowatego (spoidła wielkiego mózgu) – niepełne wykształcenie ciała modzelowatego, czyli struktury odpowiadającej za przekazywanie informacji pomiędzy półkulami mózgowymi

Zaburzenia neurologiczne u osób z zespołem Edwardsa
- obniżone napięcie mięśniowe (hipotonia) w okresie niemowlęctwa, a wzmożone napięcie mięśniowe (hipertonia) w późniejszym okresie życia
- padaczka u ok. 64% chorych z trisomią 18 (ataki drgawek w większości udaje się dobrze kontrolować przy pomocy farmakoterapii)
Wady układu moczowo-płciowego u osób z trisomią 18. chromosomu
- nerka podkowiasta, agenezja nerek (jedno- lub obustronny brak nerki), wodonercze
- u chłopców może występować: wnętrostwo, spodziectwo, mikropenis,
- u dziewczynek do objawów trisomii 18 może należeć dysgenezja (nieprawidłowy rozwój) jajników, niedorozwój warg sromowych większych, przerost łechtaczki, wady budowy macicy
Wady narządu wzroku w trisomii 18
- zaćma
- zmętnienie rogówki
- zaburzenia pigmentacji siatkówki
- malformacje naczyniowe (nieprawidłowe połączenia lub budowa naczyń krwionośnych)
- zaburzenia ostrości widzenia, nadwrażliwość na światło
U osób z zespołem Edwardsa obserwuje się również zwiększone ryzyko rozwoju nowotworu nerki (guza Wilmsa, inaczej nerczaka zarodkowego) oraz nowotworu wątroby – hepatoblastomy (inaczej wątrobiaka zarodkowego).
Diagnostyka zespołu Edwardsa
Diagnostyka trisomii 18 przebiega tak, jak diagnostyka w kierunku zespołu Downa (TRISOMIA 21 – ZESPÓŁ DOWNA).
W pierwszym trymestrze ciąży wykonywane są nieinwazyjne badania prenatalne, czyli:
- USG prenatalne pierwszego trymestru ciąży – ocenia m.in. rozwój anatomiczny płodu, a także ryzyko wystąpienia trzech trisomii w tym właśnie trisomii 18 ( Zespołu Edwardsa). Ryzyko oblicza się na podstawie daty urodzenia przyszłej mamy, wyselekcjonowanych parametrów ultrasonograficznych anatomii płodu, pomiarów płodu, ilości uderzeń serca płodu oraz markerów ultrasonograficznych zaburzeń genetycznych. Głównym markerem jest przezierność karkowa – NT, a jako dodatkowe mogą być wykorzystane: ocena obecności kości nosowej (NB), ocena przepływu w przewodzie żylnym (DV) czy też ocena przepływu przez zastawkę trójdzielną (TR)
Na temat badania usg I trymestru przeczytasz w artykule USG PRENATALNE PIERWSZEGO TRYMESTRU.

- test podwójny (tzw. test PAPP-A) – ocenia stężenie białka ciążowego A (tzw. białka PAPP-A) oraz wolnej podjednostki beta-hCG). Na temat testu przeczytasz w artykule- TEST PODWÓJNY CZYLI TEST PAPP-A.
- NIPT – nieinwazyjne testy prenatalne (np. Harmony ,Nace, Neobona, Nifty, Nifty Pro, Panorama, Sanco, Veragene, Veracity), które oceniają ryzyko wystąpienia wad genetycznych u płodu poprzez badanie wolnego płodowego DNA (cffDNA, ang. cell free fetal DNA), które znajduje się w krwi kobiety będącej w ciąży
Opierając się na uzyskanych wynikach badania USG oraz testu podwójnego oraz uwzględniając wiek kobiety będącej w ciąży lekarz wykorzystując specjalny algorytm może określić jakie jest ryzyko urodzenia dziecka z zespołem Edwardsa. Bardzo ważne jest to, że skuteczność oceny ryzyka trisomii 18 (ale również trisomii 21 i 13) wzrasta gdy analizowane są równocześnie wszystkie wymienione powyżej parametry. Wyniki powyższych przesiewowych nieinwazyjnych badań prenatalnych określają jakie jest prawdopodobieństwo urodzenia dziecka z trisomią 18 (oraz trisomią 21 i 13). Na ich podstawie nie stawia się diagnozy. Zgodnie z rekomendacjami Zespołu ekspertów Polskiego Towarzystwa Ginekologicznego oraz Polskiego Towarzystwa Genetyki Człowieka (2017), ryzyko obliczone na podstawie zintegrowanych wyników USG prenatalnego i testu podwójnego, które wynosi < 1:1000 oceniane jest jako niskie ryzyko urodzenia dziecka z trisomią 18 (oraz 21 i 13).
Jeżeli ryzyko wyliczone na podstawie wyniku badania przesiewowego I trymestru ciąży wynosi powyżej 1:1000, wskazana jest dalsza diagnostyka. W takiej sytuacji można zdecydować się na wykonanie nieinwazyjnych przesiewowych badań genetycznych wykonywanych na wolnym, płodowym DNA, tzw. testach NIPT. Należy jednak podkreślić, że testy NIPT są badaniami przesiewowymi, a więc również oceniają ryzyko wystąpienia trisomii i na ich podstawie nie można postawić diagnozy. Badania te są bezpieczne, do ich wykonania potrzebna jest próbka krwi kobiety ciężarnej, dlatego też część rodziców się na nie decyduje, gdyż stanowią dodatkowe, cenne źródło informacji o stanie zdrowia dziecka i niejednokrotnie pomagają rodzicom podjąć decyzje o wykonaniu inwazyjnych badań prenatalnych, na podstawie których można postawić ostateczną diagnozę. Do takich badań należy amniopunkcja, która jest zalecana szczególnie wtedy, gdy ryzyko wyliczone w teście zintegrowanym wynosi > 1:100.
O aminopunkcji przeczytasz więcej w artykule – AMINOPUNKCJA.

Dzięki inwazyjnym badaniom prenatalnym można określić kariotypu płodu (KARIOTYP). W badaniu ocenia się pod kątem jakościowym i ilościowym zestaw chromosomów dziecka, a na podstawie takiego wyniku można postawić diagnozę.
Kariotyp podawany jest w formie zapisu:
- kariotyp prawidłowy – zapis dla odpowiednio chłopca i dziewczynki to: 46XY lub 46XX
- zespół Edwardsa – zapis kariotypu dla odpowiednio chłopca i dziewczynki wygląda następująco 47XY,+18 lub 47XX,+18, co oznacza, że zestaw chromosomów posiada dodatkowy chromosom (zamiast 46, jest ich 47) z zaznaczeniem, który z chromosomów jest nadmiarowy (+18 – czyli chromosom 18)
Po urodzeniu dziecko z zespołem Edwardsa wymaga wielokierunkowej diagnostyki i opieki medycznej. Wsparcie poszczególnych specjalistów jest uzależnione od stanu zdrowia i potrzeb zdrowotnych dziecka.
Leczenie trisomii 18
Zespół Edwardsa to choroba, której jak innych chorób genetycznych, nie potrafimy skutecznie leczyć. Ponieważ jest to zespół wielu wad i schorzeń, osoba z trisomią 18 wymaga wielospecjalistycznej opieki. Wszystkie działania terapeutyczne są dostosowane do stanu zdrowia danej osoby. Chory jest pod opieką lekarza POZ (pediatry, lekarza rodzinnego) oraz zespołu specjalistów, jak chociażby kardiologa, neurologa, nefrologa czy fizjoterapeuty. Prowadzone leczenie ma na celu zmniejszenie objawów i poprawę jakości życia chorego. W przypadku niektórych wad wrodzonych podejmowane są zabiegi chirurgiczne, jednak każdorazowo takie decyzje podejmowane są indywidualnie w zależności od stanu zdrowia danej osoby. Czasem takie zabiegi są zbyt ryzykowne i niebezpieczne. W przypadku osób z zespołem Edwardsa zwraca się również uwagę na ważną rolę badań przesiewowych i diagnostycznych w kierunku nowotworów nerek i wątroby, których ryzyko wystąpienia u tych chorych jest wyższe niż dla ogółu populacji.
Rokowanie w zespole Edwardsa
Rokowania w przypadku zespołu Edwardsa są złe. Długość życia zależy od ilości wad i stopnia ich nasilenia. Niestety wskutek powikłań wynikających z obecności ciężkich wad wrodzonych tylko około połowa dzieci urodzonych z trisomią 18 żyje dłużej niż 1 tydzień, a jedynie 5-13,5% przeżywa powyżej roku. Dzieci, które przeżyły pierwszy rok mają 80% szans na przeżycie swoich piątych urodzin. W związku z licznymi ciężkimi objawami dzieci z trisomią 18 wymagają stałej opieki. Noworodki z zespołem Edwardsa mają problem z przyjmowaniem pokarmu. W związku z bardzo słabo wykształconym lub brakiem odruchu ssania, nie są one w stanie samodzielnie ssać pierś mamy, czy też przyjmować mleko z butelki. Osoby z trisomią 18 mają również problemy z połykaniem, krztuszą się i wymiotują podczas karmienia. Przyczynia się do tego m.in. słabo rozwinięty odruch połykania oraz wrodzona wiotkość krtani, tchawicy, przełyku i oskrzeli. Ponadto często występuje u nich silny refluks żołądkowo-przełykowy. Dlatego też niejednokrotnie jedynym rozwiązaniem jest karmienie ich przez sondę wprowadzoną do żołądka lub dojelitowo. A z kolei osłabienie mięśniówki jelit wiąże się z problemami z wypróżnianiem i częstymi zaparciami. Wady układu oddechowego, nerwowego oraz wady serca powodują, że dzieci z trisomią 18 często rozwijają nadciśnienie płucne, borykają się z niewydolnością oddechową oraz centralnym i obturacyjnym bezdechem sennym. Trisomia 18 wiąże się również ze znacznym opóźnieniem i zaburzeniami rozwoju psychoruchowego. W większości przypadków dzieci wymagają pomocy w poruszaniu się, nieliczne starsze dzieci są w stanie poruszać się przy użyciu chodzika, a w pojedynczych znanych przypadkach rozwój układu ruchu pozwolił na samodzielne poruszanie się. W większości przypadków osoby z zespołem Edwardsa są niepełnosprawne intelektualnie, najczęściej w stopniu umiarkowanym lub ciężkim.
Bibliografia
- Balasundaram P, Avulakunta ID. Edward Syndrome. 2021 Jun 10. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan–. PMID: 34033359.
- Cuckle H, Morris J. Maternal age in the epidemiology of common autosomal trisomies. Prenat Diagn. 2021 Apr;41(5):573-583. doi: 10.1002/pd.5840. Epub 2020 Oct 19. PMID: 33078428.
- Kevin L Howe, Premanand Achuthan, James Allen, Jamie Allen, Jorge Alvarez-Jarreta, M Ridwan Amode, Irina M Armean, Andrey G Azov, Ruth Bennett, Jyothish Bhai, Konstantinos Billis, Sanjay Boddu, Mehrnaz Charkhchi, Carla Cummins, Luca Da Rin Fioretto, Claire Davidson, Kamalkumar Dodiya, Bilal El Houdaigui, Reham Fatima, Astrid Gall, Carlos Garcia Giron, Tiago Grego, Cristina Guijarro-Clarke, Leanne Haggerty, Anmol Hemrom, Thibaut Hourlier, Osagie G Izuogu, Thomas Juettemann, Vinay Kaikala, Mike Kay, Ilias Lavidas, Tuan Le, Diana Lemos, Jose Gonzalez Martinez, José Carlos Marugán, Thomas Maurel, Aoife C McMahon, Shamika Mohanan, Benjamin Moore, Matthieu Muffato, Denye N Oheh, Dimitrios Paraschas, Anne Parker, Andrew Parton, Irina Prosovetskaia, Manoj P Sakthivel, Ahamed I Abdul Salam, Bianca M Schmitt, Helen Schuilenburg, Dan Sheppard, Emily Steed, Michal Szpak, Marek Szuba, Kieron Taylor, Anja Thormann, Glen Threadgold, Brandon Walts, Andrea Winterbottom, Marc Chakiachvili, Ameya Chaubal, Nishadi De Silva, Bethany Flint, Adam Frankish, Sarah E Hunt, Garth R IIsley, Nick Langridge, Jane E Loveland, Fergal J Martin, Jonathan M Mudge, Joanella Morales, Emily Perry, Magali Ruffier, John Tate, David Thybert, Stephen J Trevanion, Fiona Cunningham, Andrew D Yates, Daniel R Zerbino, Paul Flicek, Ensembl 2021, Nucleic Acids Research, Volume 49, Issue D1, 8 January 2021, Pages D884–D891.
- Outtaleb FZ, Errahli R, Imelloul N, Jabrane G, Serbati N, Dehbi H. La trisomie 18 ou syndrome d’Edwards en post-natal: étude descriptive au Centre Hospitalier Universitaire de Casablanca et revue de littérature [Trisomy 18 or postnatal Edward´s syndrome: descriptive study conducted at the University Hospital Center of Casablanca and literature review]. Pan Afr Med J. 2020 Dec 3;37:309. French. doi: 10.11 Meyer RE, Liu G, Gilboa SM, Ethen MK, Aylsworth AS, Powell CM, Flood TJ, Mai CT, Wang Y, Canfield MA; National Birth Defects Prevention Network. Survival of children with trisomy 13 and trisomy 18: A multi-state population-based study. Am J Med Genet A. 2016 Apr;170A(4):825-37. doi: 10.1002/ajmg.a.37495. Epub 2015 Dec 10. PMID: 26663415; PMCID: PMC4898882.604/pamj.2020.37.309.26205. PMID: 33654528; PMCID: PMC7896527.
- Peron A., Carey J.C. The Molecular Genetics of Trisomy 18: Phenotype–Genotype Correlations. 14 November 2014 https://doi.org/10.1002/9780470015902.a0025246. eLS & 2014, John Wiley & Sons, Ltd.
- Rosa RF, Rosa RC, Zen PR, Graziadio C, Paskulin GA. Trisomy 18: review of the clinical, etiologic, prognostic, and ethical aspects. Rev Paul Pediatr. 2013 Jan-Mar;31(1):111-20. English, Portuguese. doi: 10.1590/s0103-05822013000100018. PMID: 23703053
- Cereda A, Carey JC. The trisomy 18 syndrome. Orphanet J Rare Dis. 2012 Oct 23;7:81. doi: 10.1186/1750-1172-7-81. PMID: 23088440; PMCID: PMC3520824.
- Nusbaum C, Zody MC, Borowsky ML et al. DNA sequence and analysis of human chromosome 18. Nature 2005; 437(7058): 551–555
- Rasmussen SA, Wong LY, Yang Q, May KM, Friedman JM. Population-based analyses of mortality in trisomy 13 and trisomy 18. Pediatrics. 2003 Apr;111(4 Pt 1):777-84. doi: 10.1542/peds.111.4.777. PMID: 12671111.
- Boghosian-Sell L, Mewar R, Harrison W, Shapiro RM, Zackai EH, Carey J, Davis-Keppen L, Hudgins L, Overhauser J. Molecular mapping of the Edwards syndrome phenotype to two noncontiguous regions on chromosome 18. Am J Hum Genet. 1994 Sep;55(3):476-83. PMID: 8079991; PMCID: PMC1918415.
- EDWARDS JH, HARNDEN DG, CAMERON AH, CROSSE VM, WOLFF OH. A new trisomic syndrome. Lancet. 1960 Apr 9;1(7128):787-90. doi: 10.1016/s0140-6736(60)90675-9. PMID: 13819419.
- SMITH DW, PATAU K, THERMAN E, INHORN SL. A new autosomal trisomy syndrome: multiple congenital anomalies caused by an extra chromosome. J Pediatr. 1960 Sep;57:338-45. doi: 10.1016/s0022-3476(60)80241-7. PMID: 13831938.
- Hook EB. Rates of chromosome abnormalities at different maternal ages. Obstet Gynecol 1981 , 58, 282-5. PMID: 6455611
- Hook EB, Cross PK & Schreinemachers DM. Chromosomal abnormality rates at amniocentesis and in live-born infants. JAMA 1983 , 249, 2034-8. PMID: 6220164
- Schreinemachers DM, Cross PK & Hook EB. Rates of trisomies 21, 18, 13 and other chromosome abnormalities in about 20 000 prenatal studies compared with estimated rates in live births. Hum. Genet. 1982 , 61, 318-24. PMID: 6891368
- Rekomendacje Zespołu Ekspertów Polskiego Towarzystwa Ginekologicznego oraz Polskiego Towarzystwa Genetyki Człowieka w zakresie przesiewowego badania genetycznego wykonanego na wolnym płodowym DNA, Ginekologia i Perinatologia Praktyczna 2017, tom 2, nr 5, 230–233.
- Rekomendacje Sekcji Ultrasonografii Polskiego Towarzystwa Ginekologów i Położników w zakresie przesiewowej diagnostyki ultrasonograficznej w ciąży o przebiegu prawidłowym – 2020 r.